PROTAC Design Guide: Synthesis Strategy for VHL PROTAC Linker
Abstract
VHL, with its widespread expression across tissue types, has become one of the most extensively used E3 ligases in PROTAC development. It has been demonstrated that the composition and length of the linker play a crucial role in influencing the spatial orientation and physicochemical properties of the POI-PROTAC-E3 ternary complex. In this review, the authors focus on providing an overview of the synthesis strategies for the linker component in current VHL PROTAC compositions.
Fig 1. Targeted protein degradation catalyzed by PROTACs
Introduction
PROTACs, or Proteolysis Targeting Chimeras, consist of three components: a ligand for the target protein (POI), a ligand for the E3 ubiquitin ligase, and a linker that connects the two parts. PROTACs hijack the cell’s natural ubiquitin-proteasome system (UPS) to recruit various E3 ligases and induce protein degradation. However, due to the high molecular weight and poor physicochemical properties of PROTACs, many of them suffer from issues such as poor cellular uptake or pharmacokinetics/pharmacodynamics (PK/PD). Modulating the linker represents a suitable optimization approach to achieve the correct combination of length, hydrophilicity, and rigidity in order to address these challenges effectively.
An effective approach is to generate a PROTACs database containing linear alkyl linkers of varying lengths until the appropriate length and orientation between the POI and E3 ligase can be identified. Currently, flexible linkers ranging from a few atoms to 29 atoms in length are commercially available. Flexible, straight-chain alkyl, and PEG linkers are increasingly being replaced by more rigid structures (e.g., alkynes) and cyclic scaffolds (e.g., piperazine, pyridine, triazole), providing opportunities to modulate PROTAC activity and various properties such as hydrophilicity/hydrophobicity, metabolic stability, bioavailability, and cell membrane permeability.
Fig 2. Representative examples of VHL PROTACs
The general method of introducing Linker in VHL-PROTAC
VHL is part of the E3 ubiquitin ligase complex CUL2-RBX1-ElonginB-ElonginC-VHL, consisting of two structural domains with specific binding sites. It recruits hypoxia-inducible factor 1 alpha (HIF-1A) and induces its ubiquitination followed by proteasomal degradation.
To date, VHL-based PROTACs have been successfully used to degrade over 20 target proteins. Following the discovery of the first small-molecule VHL inhibitor, VH032 (1) (Kd = 185 nM), modifications around its acetamide group led to the development of second-generation VHL inhibitors VH101 (2) (Kd = 44 nM) and VH298 (3) (Kd = 90 nM) with four-fold higher potency. Building upon these mature VHL inhibitors, a range of small molecule VHL ligands suitable for linker connection have been developed to generate VHL-recruiting PROTACs (Fig 2).
So far, over 1500 different linkers have been reported in PROTAC design (data from PROTAC-DB 2.0; http://cadd.zju.edu.cn/protacdb/). Traditionally, three methods have been employed in assembling VHL-based PROTACs:
Method A: Linker is first attached to the VHL ligand and then connected to the POI warhead.
Method B: Linker is first attached to the POI warhead and then connected to the VHL ligand.
Method C: Two smaller adapter fragments are installed on the POI and E3 ligand, respectively, followed by their connection.
Optimizing the linker design involves considering various factors such as length, flexibility, hydrophilicity/hydrophobicity, and stability. The choice of linker strategy depends on the specific requirements of the target protein and E3 ligase interaction, as well as the desired pharmacokinetic properties of the PROTAC molecule. The availability of a diverse library of linkers offers opportunities for exploring and optimizing PROTAC designs to achieve enhanced efficacy and target specificity.
Flexible Linker for VHL-PROTAC
Flexible linkers typically consist of straight-chain alkyl and polyethylene glycol (PEG) units and are the most common type of linker found in PROTAC literature. The presence of numerous rotatable bonds provides PROTACs with greater flexibility, allowing for a larger number of low-energy conformations in solution. This “casting a wide net” approach enables the exploration of diverse conformations, maximizing the potential for formation of ternary complexes with the POI and E3 ligase.
Due to the improved hit rate in forming ternary complexes and subsequent degradation of the POI, flexible linkers are often the preferred choice in the early stages of PROTAC discovery. The conformational freedom imparted by flexible linkers also allows PROTACs to collapse into compact conformations in polar media such as plasma or cytoplasm. This reduces the effective polar surface area of the compound and can shield certain hydrogen donors and acceptors through intramolecular hydrogen bonding, thereby improving permeability and oral bioavailability.
For example, a one-atom change demonstrated by C4 at this year’s AACR conference led to the collapse of the PROTAC conformation, resulting in a significant increase in oral bioavailability by several folds.
Hence, experimental determinations of ADME properties may significantly differ from computer calculations. In particular, it is strongly recommended to use experimentally determined effective polar surface area (EPSA) rather than TPSA.
Based on the authors’ experience, the chemical reactions used to incorporate flexible adapters into PROTACs are robust and consistently perform well regardless of linker length or type. This enables the use of a universal method for constructing PROTAC libraries. Typical conditions for linker attachment to the VHL ligand are shown in Table 1.
Table 1. Common synthetic methods for linking Linker to VHL ligands.
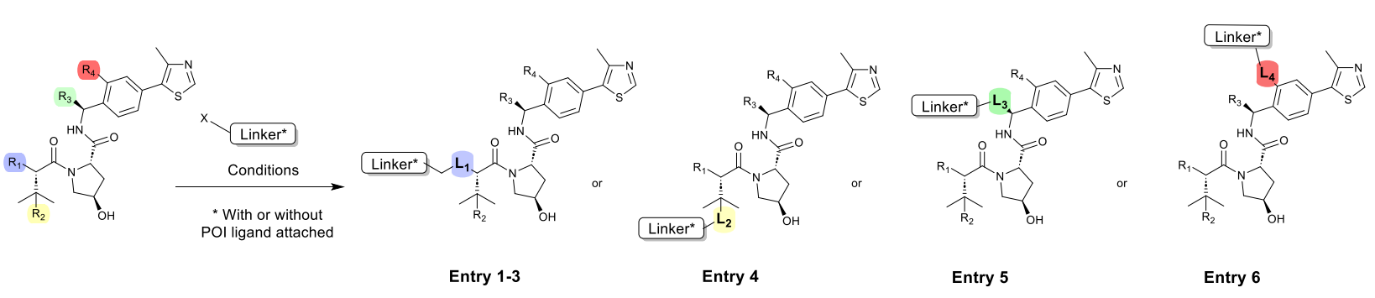
The most common method for linker attachment is the formation of an amide bond between the terminal amine of the VHL ligand and the linker (1). Alternative approaches include the use of secondary amines through reductive amination or alkylation (2 and 3). The use of thioethers is less common (4). Another increasingly popular approach involves using a connection at position 3 attached to a benzyl group (5).
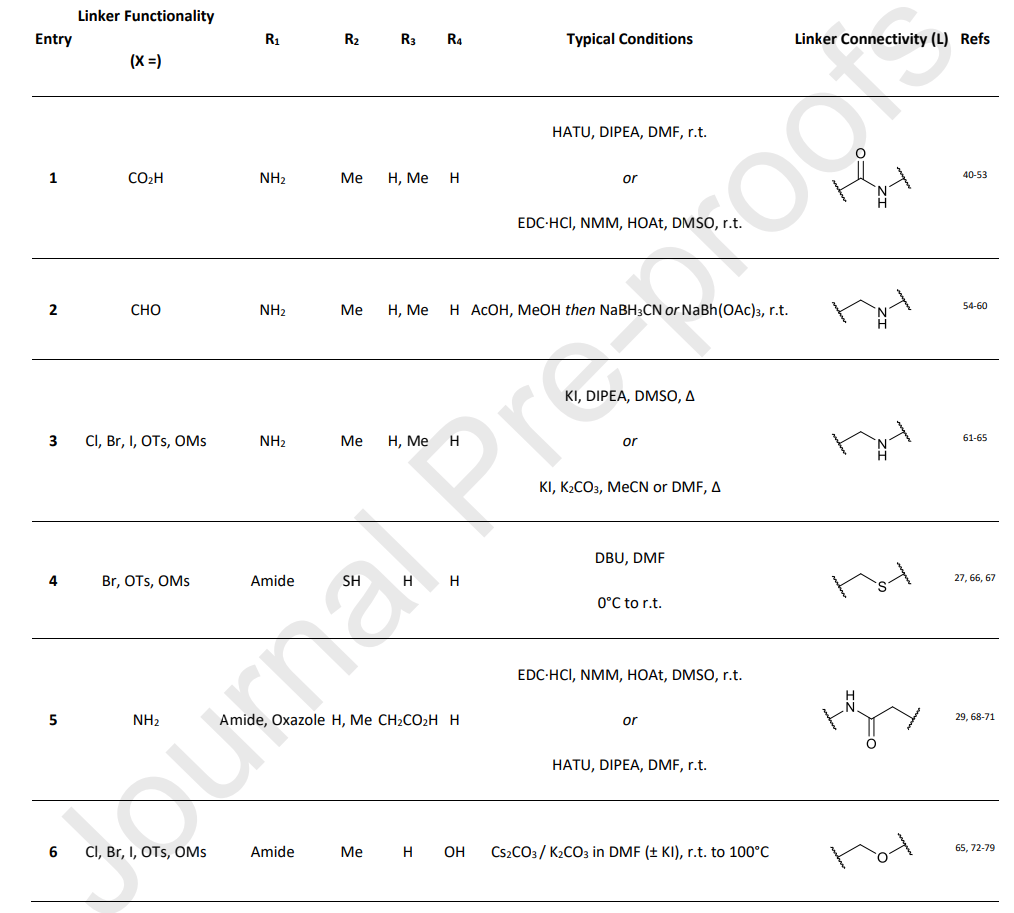
In addition to amide bonds at the benzyl position of the VHL ligand, PROTACs containing benzyl ether and carbon-carbon bonds have been reported. The work of Kofink et al. serves as an example, where these novel benzyl bond types were utilized to develop selective degraders of SMARCA2, such as compound 15 (Fig 3).
Fig 3. SMARCA2 degrader of carbon-tethered benzyl linker
The solvent exposure properties of the 4-methyl substituent on the thiazole ring were identified early in VHL ligand SAR studies. Derivatization at this position has been assumed in patent literature. However, the first reported example of a thiazole-linked VHL PROTAC was recently described by Krieger et al. from Merck & Co.
While flexible linkers have been the most commonly used adapters in VHL-targeting PROTACs to date, many optimized PROTACs employ more rigid and functionalized linkers rather than simple linear chains. This is partly because rigid linkers can achieve higher potency, biasing the conformation of PROTACs towards the biologically active conformations observed in the ternary complex in solution.
Moreover, linker-protein interactions can play a crucial role in the stability of the POI-PROTAC-E3 ternary complex. Therefore, the use of more specialized linkers can better exploit interactions with surface residues of both the POI and E3 proteins, rather than relying on simple PEG or alkyl chains.
As PROTACs move towards in vivo studies, the metabolic sensitivity of PEG chains is a particular concern. On the other hand, using straight-chain alkyl linkers can result in poor water solubility, especially in VHL-targeting PROTACs where the E3 ligand often exhibits hydrophobicity compared to CRBN ligands, ultimately leading to reduced bioavailability of PROTACs.
Rigid Linker for VHL-PROTAC
Once the optimal linker length is determined through biological evaluation or computational prediction using a linear flexible linker, the introduction of a similarly sized rigid linker follows. Common rigid motifs in PROTAC linker design include (hetero)cycles, alkynes, and spirolactones, which account for 37%, 3.4%, and 1.1% of the reported PROTACs, respectively (Fig 4).
Incorporating rigid linkers can be advantageous for enhancing water solubility, cell permeability, and improving the pharmacokinetic properties of degraders. Due to these potential benefits, many PROTACs in clinical trials feature short yet highly rigid adapters.
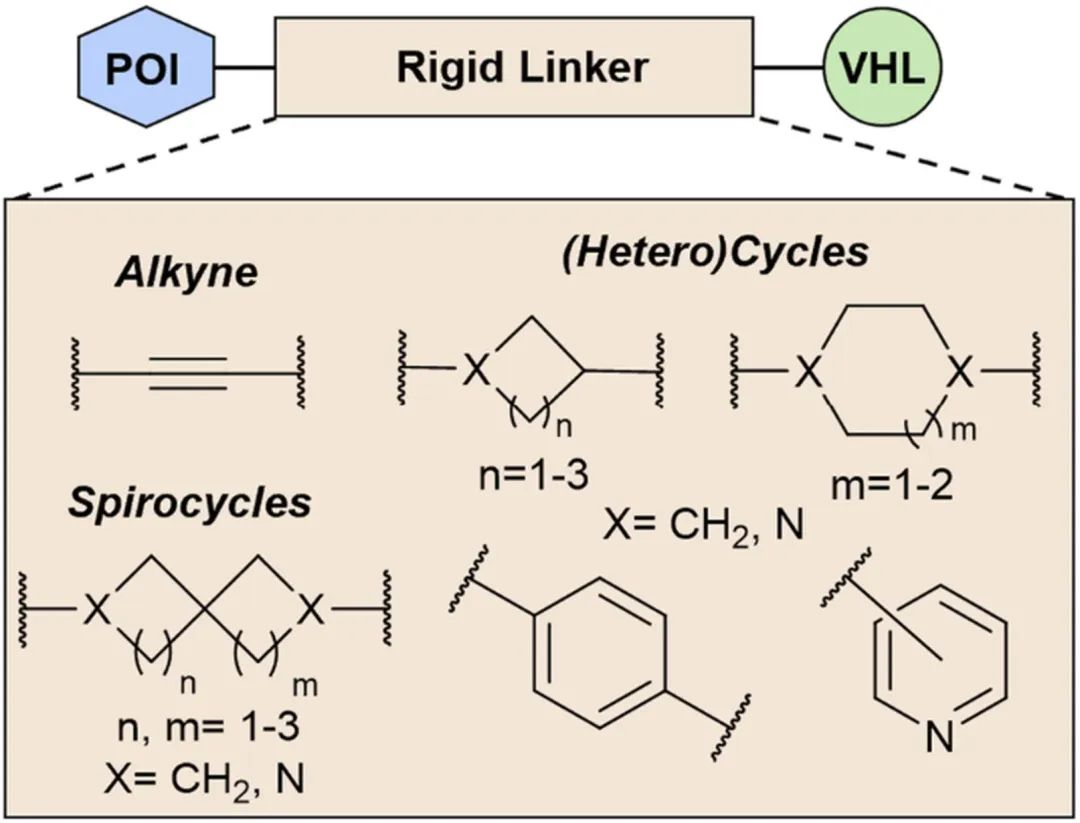
Fig 4. Key Rigid Linker
Replacing a linear chain with a rigid scaffold reduces the number of rotatable bonds and directs the entire conformation of the POI-E3 ligase ternary complex in a constrained manner, potentially improving protein degradation. Wang’s work serves as an example, where they developed and evaluated a series of AR degraders through extensive optimization around the linker. Substituting the flexible linker with a rigid one resulted in a 76% increase in degradation potency at 0.01 μM.
Interestingly, the Ciulli group also explored macrocyclic linkers as an alternative strategy to lock the BET degrader MZ1 (21) into a conformation close to its active state. Crystal structures suggested the placement of a second linker between the benzene ring of the VHL ligand and the first PEG linker of MZ1 to form a macrocyclic linker. Despite a 12-fold affinity loss, the macro PROTAC (22) exhibited similar cellular potency to MZ1.
The choice of chemical reactions used to connect rigid linkers is typically determined by the functionality of the VHL ligand and the POI ligand. The introduction of rigid linkers in VHL-PROTAC design can be achieved through three methods.
The first method (Method A)
Amide coupling conditions are commonly used for PROTAC assembly (23, 24), which involves conjugating an amino-functionalized linker with a carboxylic acid-containing head group (Table 2). Another linking strategy reported by Nunes et al. for developing IRAK4 PROTACs involves pre-conjugating a linker with an alkyne moiety to the POI head group via Sonogashira cross-coupling reaction, followed by coupling with the VHL ligand (25). Reductive amination has also been employed as an alternative synthetic strategy (26).
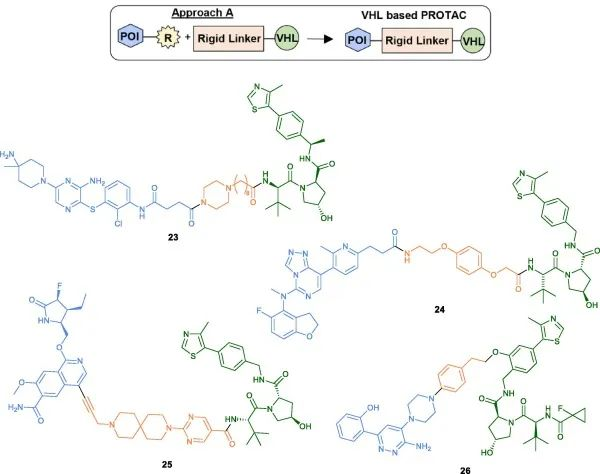
Fig 5. Examples of VHL-PROTACs synthesized using Method A
The second method (Method B)
The specific coupling conditions for the final step are typically determined by the type and respective functionalities of the VHL ligand. In addition to the commonly used amide coupling conditions (27-30), the Ciulli group recently discovered XL01126, a BBB-permeable and effective LRRK2 degrader. For the synthesis of XL01126, coupling of the linker to the VHL ligand was achieved by using a linker with a good leaving group and DBU as the base (31).
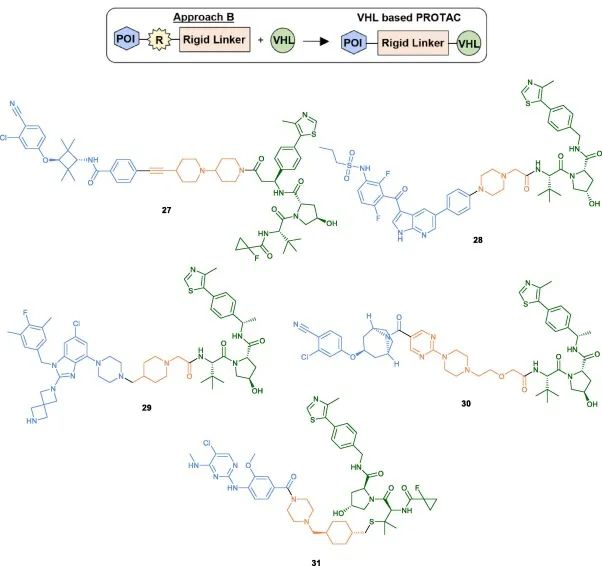
Fig 6. Examples of VHL-PROTACs synthesized using Method B
The third method (Method C)
The third method (Method C) involves initially coupling a rigid heterocycle to the POI head, followed by the final coupling to the pre-conjugated linker on the VHL ligand. A representative example is the development of an effective BCL-xL/2 dual degrader PPC8, where the linker of the VHL ligand is tethered to the pyrimidine ring of the POI head using HATU coupling (32). Bollu et al. also described a similar synthetic strategy for the synthesis of IDO1 degraders (33).
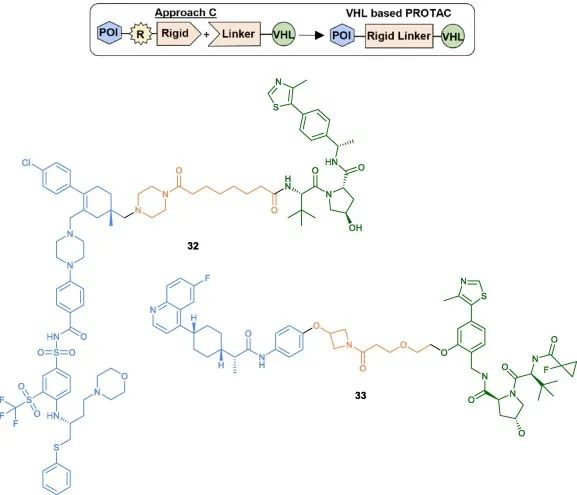
Fig 7. Examples of VHL-PROTACs synthesized using Method C
While these methods are widely used, developing PROTAC libraries with rigid linkers of varying lengths remains time-consuming. In particular, it often requires multiple synthetic steps and challenging purification processes due to the need for linker modification at multiple points. Additionally, the commercial availability of pre-assembled rigid linker-VHL ligand conjugates is limited and expensive. Having a method to rapidly expand PROTAC libraries with rigid linkers of different lengths would be invaluable.
Clickable Linkers for VHL PROTACs
Click chemistry has been widely used as a rapid method in PROTAC research to facilitate the development of various PROTAC libraries. Lebraud et al. introduced the concept of Clickable Linkers (CLIPTACs), which overcome issues related to solubility and cellular permeability by “clicking” two small-molecule PROTAC precursors inside cells. Since then, click chemistry has been recognized as a platform that enables quick access to PROTAC libraries with different linker lengths.
Copper-catalyzed Huisgen cycloaddition is one of the most common “click” platforms used for assembling the triazole moiety. This reaction occurs between azides and alkynes under mild conditions, yielding 1,4-disubstituted 1,2,3-triazoles with high yield and selectivity.
As bioisosteric replacements for amide bonds, triazoles have also been utilized to modulate the physicochemical properties of PROTACs, significantly improving their water solubility. Additionally, the triazole ring can confer rigidity to the linker and, depending on their positions on the linker, they may leverage novel intermolecular interactions, thereby enhancing the stability of the ternary complex and ultimately improving target degradation efficiency.
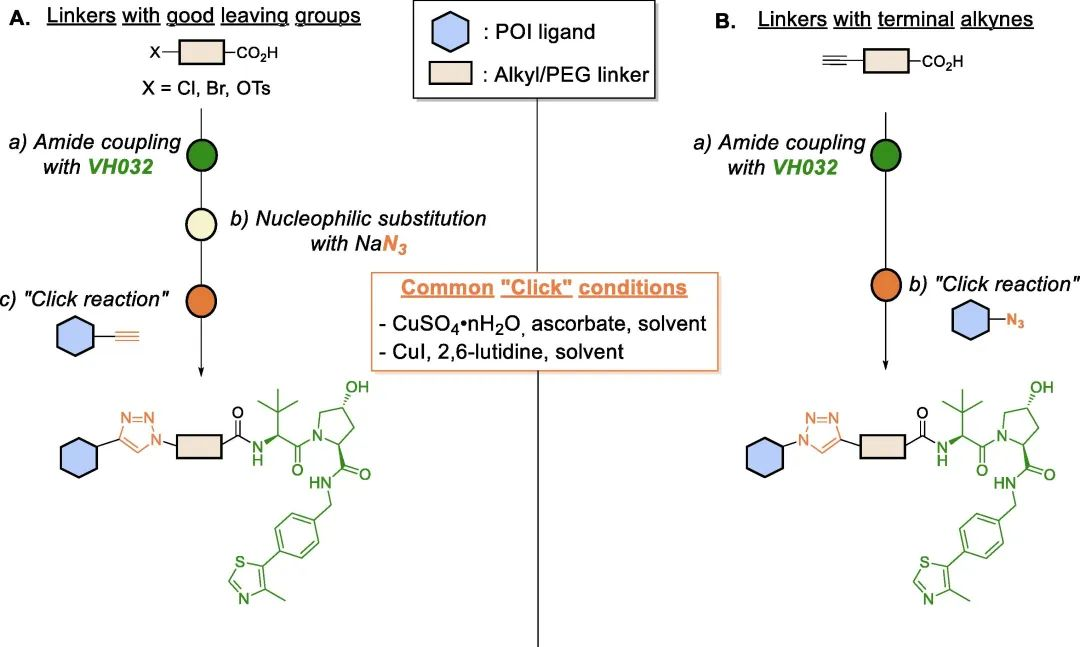
Fig 8. Schematic diagram of VHL-based PROTAC assembled by “click” chemistry
Optical Switch Linkers in VHL PROTAC
PhotoPROTACs are a novel class of degraders that combine two emerging strategies: photopharmacology and PROTACs. The concept of PhotoPROTACs was initially introduced by Crews’ group as tools for spatiotemporal control of PROTAC activity, enabling reversible on/off switching of target protein degradation. The first design of a photoPROTAC was based on the mature BET protein degrader ARV-771, which consisted of BRD4 ligand and VHL ligand connected by an 11 Å long PEG linker. The replacement of a hydrophilic moiety with an ortho-F substituted azobenzene moiety facilitated the development of the first PhotoPROTAC.
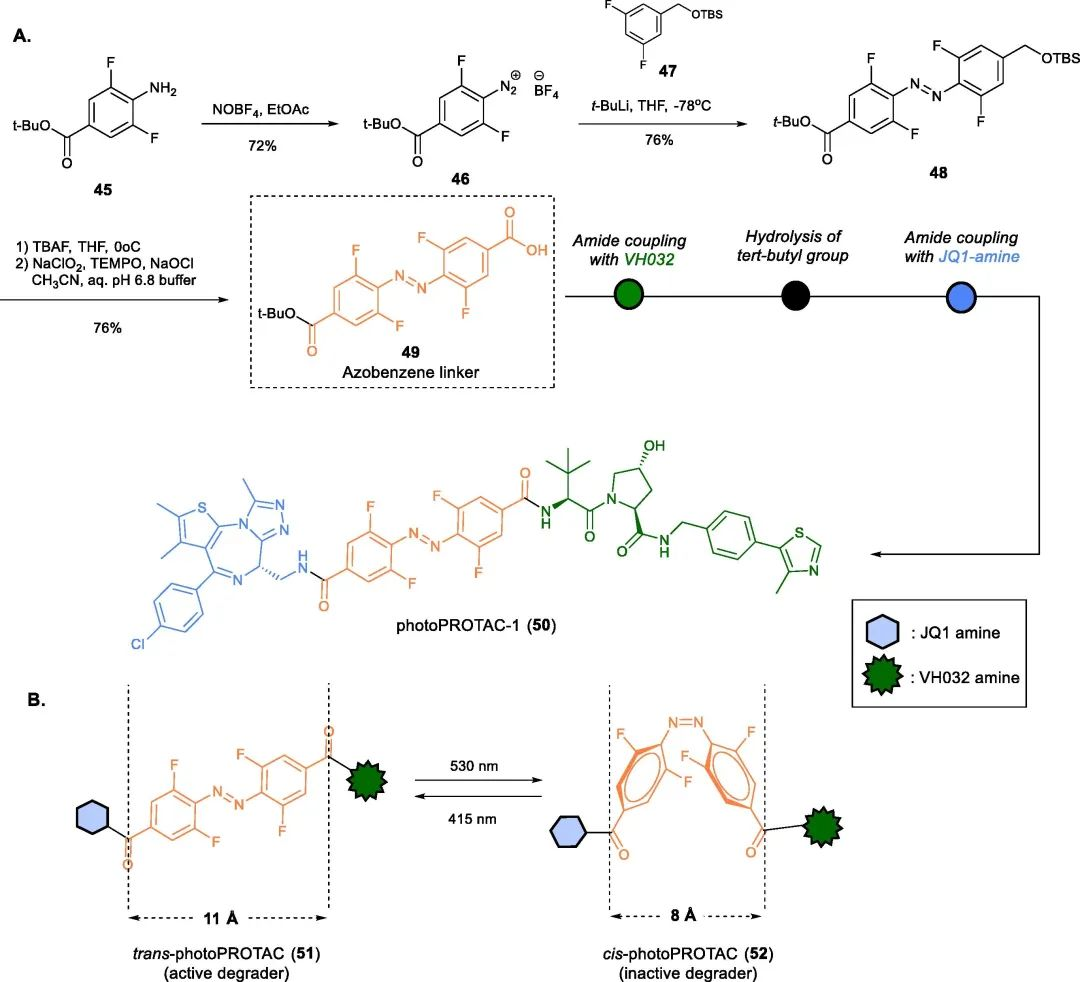
Fig 9. photoPROTACs-1(50)
Irradiation of the photoPROTAC-530 with light at 1 nm and 415 nm LEDs resulted in the formation of 95% trans-PHOTOPROTAC-1 (51) and 68% cis-PHOTOPROTAC-1 (52), respectively. Interestingly, incubation of Ramos cells with trans-PHOTOPROTAC-1 (51) induced degradation of BRD2, which continued for 17 hours, while cis-PHOTOPROTAC-1 (52) did not show any significant effect on BRD2 levels.