VEGFR-2 in Focus: Structure, Signaling, and Its Emerging Role in Disease and Therapy
Abstract
VEGFR-2 (vascular endothelial growth factor receptor 2) is a central regulator of angiogenesis, orchestrating endothelial cell proliferation, migration, survival, and vascular permeability. This blog explores the molecular structure of VEGFR-2, the signaling pathways it activates, and its critical roles in both physiological and pathological angiogenesis. Special attention is given to its involvement in diseases such as cancer, diabetic retinopathy, and rheumatoid arthritis, where VEGFR-2 signaling becomes dysregulated. Finally, the article discusses current therapeutic strategies targeting VEGFR-2, including antibody therapies, kinase inhibitors, and RNA-based approaches. Understanding VEGFR-2 not only deepens insight into vascular biology but also paves the way for innovative treatments in angiogenesis-related disorders.
Introduction to VEGFR-2 and Angiogenesis
Angiogenesis, the process of forming new blood vessels from existing vasculature, is one of the most fundamental biological mechanisms in human health and disease. From wound healing and embryonic development to tumor growth and retinal disorders, angiogenesis plays a pivotal role in shaping tissue responses to both physiological and pathological stimuli.
At the heart of this complex vascular remodeling is a family of signaling proteins known as vascular endothelial growth factors (VEGFs). Among them, VEGF-A is the most well-characterized, acting as a potent mitogen for endothelial cells. But VEGF cannot function alone—it requires specific receptors on the cell surface to transmit its signal. That’s where vascular endothelial growth factor receptor 2 (VEGFR-2) steps in.
VEGFR-2, also known as KDR or Flk-1, is the principal receptor mediating the pro-angiogenic effects of VEGF. Expressed primarily on vascular endothelial cells, VEGFR-2 initiates intracellular signaling cascades once it binds to VEGF-A. This activation leads to a cascade of biological responses including endothelial cell proliferation, migration, survival, and increased vascular permeability—all critical components of vessel sprouting and maturation.
What sets VEGFR-2 apart is its dual role in health and disease. In embryogenesis, it ensures proper blood vessel formation and supports hematopoietic development. In adults, it regulates tissue repair, organ regeneration, and reproductive function. However, this same system can go awry. In cancer, for instance, tumors hijack VEGFR-2 signaling to establish new blood vessels that nourish malignant growth. Similarly, diseases like diabetic retinopathy and rheumatoid arthritis feature abnormal VEGFR-2 activation, contributing to chronic inflammation and tissue damage.
Understanding VEGFR-2 is not just an academic exercise—it’s a clinical necessity. Therapies that target this receptor are already in use for various cancers and ocular diseases. As research continues to uncover new details about VEGFR-2’s molecular structure and signaling pathways, it opens doors to more precise and effective strategies for regulating angiogenesis in both healing and disease contexts.
VEGFR-2 Structure – A Molecular Blueprint for Function
The biological power of VEGFR-2 lies not just in its presence, but in its intricate structure. As a type V receptor tyrosine kinase, VEGFR-2 is uniquely engineered to respond to vascular endothelial growth factors (VEGFs) and convert their extracellular messages into potent intracellular actions. Its structural complexity is what enables it to serve as a master regulator of angiogenesis.
VEGFR-2 is composed of 1,356 amino acids and is organized into five major domains: the extracellular domain (ECD), transmembrane domain (TMD), juxtamembrane domain (JMD), tyrosine kinase domain (TKD), and C-terminal domain (CTD).
The extracellular domain (20–764 amino acids) contains seven immunoglobulin-like subdomains (IgD1–IgD7). Among these, IgD2 and IgD3 are essential for binding VEGF ligands. This binding prompts receptor dimerization, a necessary step for signal transduction. Interestingly, N-linked glycosylation sites scattered across the ECD enhance ligand binding and receptor stability, emphasizing the importance of post-translational modifications in VEGFR-2 function.
Once dimerized, the intracellular regions come into play. The juxtamembrane domain houses tyrosine residue Y801, a phosphorylation site critical for initiating downstream signaling. In its unphosphorylated state, this domain acts as an autoinhibitory element by physically interacting with the activation loop (A-loop) in the kinase domain. Phosphorylation at Y801 relieves this inhibition, allowing the receptor to adopt an active conformation.
The tyrosine kinase domain (TKD) is split into three sub-regions: TKD1 (ATP-binding site), the kinase insert domain (KID), and TKD2 (phosphotransferase site). Key phosphorylation sites such as Y951, Y1054, and Y1059 reside here and are responsible for activating multiple downstream pathways including PI3K-Akt and MAPK.
Finally, the C-terminal domain (CTD) features Y1175 and Y1214, two phosphorylation hotspots that bind adapter proteins like PLCγ, SHB, and NCK. These interactions drive cellular responses like migration, proliferation, and vascular permeability.
Taken together, VEGFR-2’s modular architecture ensures that a single ligand-binding event can orchestrate a diverse array of biological responses. Understanding this structure-function relationship is crucial for designing targeted therapies that disrupt pathological angiogenesis without harming normal vascular function.
Decoding the VEGFR-2 Signaling Pathways
Once vascular endothelial growth factor (VEGF) binds to VEGFR-2 and triggers receptor dimerization, the real action begins inside the cell. VEGFR-2 signaling cascades act like molecular relay races, converting an extracellular signal into a coordinated set of cellular responses—most notably, endothelial cell proliferation, migration, survival, and vascular permeability.
The key to this process lies in the tyrosine phosphorylation of specific intracellular residues on VEGFR-2. Each phosphorylated site serves as a docking station for signaling proteins, enabling the activation of multiple downstream pathways simultaneously.
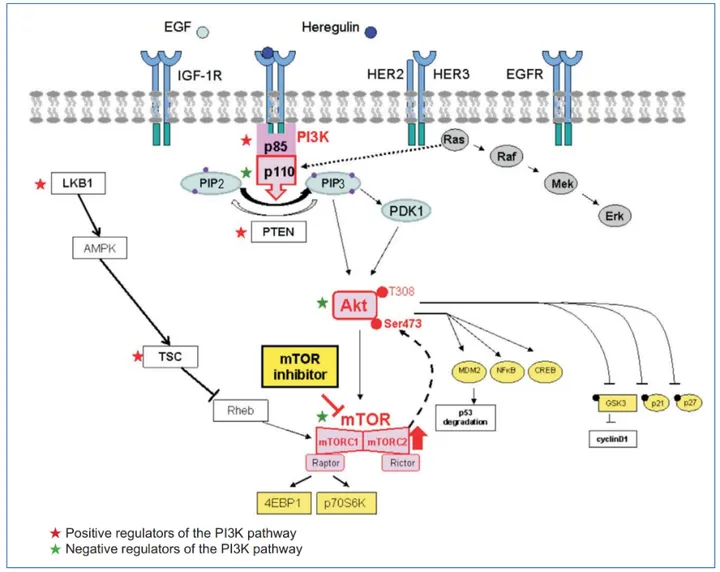
One of the most studied cascades is the PLCγ–PKC–MAPK pathway. After phosphorylation at Tyr1175, VEGFR-2 recruits and activates phospholipase C-γ (PLCγ). PLCγ then catalyzes the breakdown of PIP2 into diacylglycerol (DAG) and inositol triphosphate (IP3), leading to PKC activation and increased intracellular calcium. This ultimately triggers the MAPK/ERK pathway, which enters the nucleus and promotes DNA synthesis and endothelial cell proliferation.
Meanwhile, Tyr951 phosphorylation initiates the TSAd-Src-PI3K-Akt pathway, which is pivotal for cell survival. PI3K catalyzes the conversion of PIP2 to PIP3, activating Akt (protein kinase B), a well-known regulator of anti-apoptotic processes. Akt phosphorylates and inactivates pro-apoptotic proteins like BAD and caspase-9, ensuring the longevity of endothelial cells under VEGF stimulation.
For cell migration, two pathways stand out. Phosphorylated Tyr1175 also activates SHB, an adapter protein that links to FAK and paxillin, modulating cytoskeletal reorganization. Simultaneously, Tyr1214 phosphorylation recruits NCK, which activates the p38-MAPKAPK2/3 cascade through small GTPases like Rac and Cdc42. These events lead to dynamic actin remodeling and directed endothelial movement—critical steps in vessel sprouting.
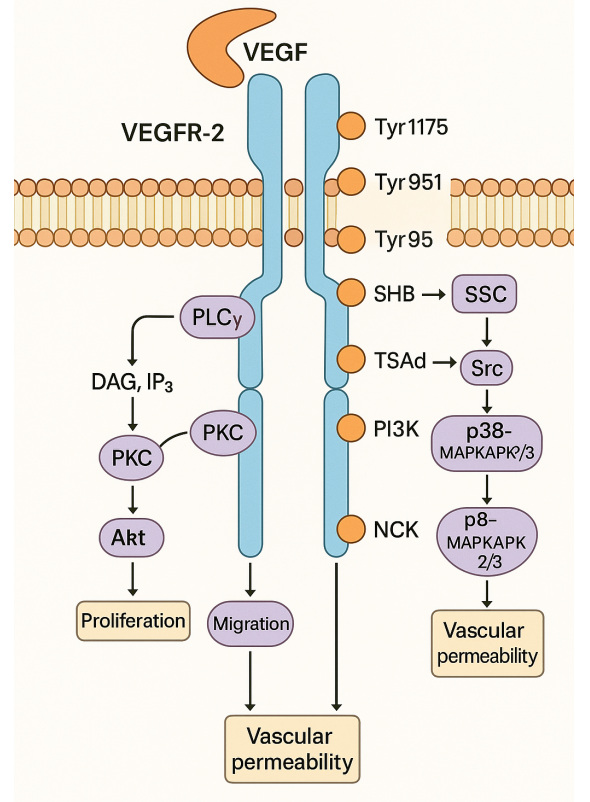
Fig. 1. VEGFR-2 Signaling Pathways
The Try801 site, located in the juxtamembrane domain, supports both proliferation and vascular permeability by activating eNOS and facilitating nitric oxide (NO) production, which relaxes blood vessels and increases permeability.
In sum, VEGFR-2 orchestrates an elegant and highly regulated network of signaling pathways. Each pathway is like a branch stemming from a central trunk, tailored to meet the specific demands of angiogenesis. Dissecting these pathways has been key to understanding—and therapeutically targeting—abnormal vessel growth in disease.
When Things Go Wrong – VEGFR-2 in Disease
While VEGFR-2 is indispensable for healthy blood vessel formation, its dysregulation is a driving force behind numerous diseases, particularly those involving aberrant angiogenesis. Under normal conditions, VEGFR-2 activation is tightly controlled to support physiological functions such as embryonic development, wound healing, and tissue regeneration. But when its signaling goes unchecked or is hijacked by pathological processes, VEGFR-2 becomes a key contributor to disease progression.
One of the most well-documented roles of VEGFR-2 dysfunction is in cancer. Tumors exploit VEGFR-2-mediated angiogenesis to create their own blood supply, fueling unchecked growth and metastasis. Cancer cells secrete high levels of VEGF, which binds to VEGFR-2 on surrounding endothelial cells. This initiates vessel sprouting toward the tumor, ensuring a continuous delivery of oxygen and nutrients. The resulting vasculature, however, is often abnormal—leaky, disorganized, and poorly functional—further complicating treatment and promoting tumor dissemination.
VEGFR-2 is also heavily implicated in ocular diseases like diabetic retinopathy and age-related macular degeneration (AMD). In these conditions, VEGF expression is upregulated in response to hypoxia or oxidative stress, leading to excessive VEGFR-2 activation and pathological neovascularization in the retina. These fragile new vessels often rupture, causing bleeding, edema, and eventual vision loss.
Beyond oncology and ophthalmology, VEGFR-2 plays a significant role in chronic inflammatory diseases such as rheumatoid arthritis. Inflammatory cells release pro-angiogenic cytokines and VEGF, activating VEGFR-2 and promoting synovial angiogenesis. This sustains inflammation and joint destruction by enabling continuous immune cell infiltration.
Furthermore, research links VEGFR-2 to neurodegenerative conditions like Alzheimer’s disease, where aberrant angiogenesis and blood-brain barrier breakdown may exacerbate neuronal damage. VEGFR-2-mediated permeability increases vascular leakage, allowing toxic proteins and inflammatory factors to accumulate in the brain.
Altogether, these findings highlight VEGFR-2 as a double-edged sword—vital for healing, yet dangerous when overactivated. Its involvement in such a wide range of diseases makes it a prime target for therapeutic intervention, as explored further in the next section.
Targeting VEGFR-2 – A Strategy for Therapeutic Innovation
Given VEGFR-2’s central role in angiogenesis and its implication in a wide range of diseases, it has emerged as a prime therapeutic target—especially in cancer, ocular disorders, and chronic inflammatory diseases. VEGFR-2-targeted therapies aim to disrupt the VEGF/VEGFR-2 signaling cascade, halting the pathological growth of blood vessels while sparing healthy vasculature.
One of the most effective strategies involves blocking VEGF from binding to VEGFR-2. This can be accomplished using neutralizing antibodies like bevacizumab (Avastin), which sequester VEGF and prevent receptor activation. Another approach uses soluble VEGFR-2 decoy receptors (e.g., aflibercept) that act as molecular sponges, trapping VEGF before it reaches the endothelial cell surface.
A second strategy targets VEGFR-2 directly at the receptor level, using small-molecule tyrosine kinase inhibitors (TKIs). Drugs like sunitinib, sorafenib, and axitinib bind to the intracellular kinase domain of VEGFR-2, preventing its phosphorylation and downstream signaling. These oral agents have shown success in treating renal cell carcinoma, hepatocellular carcinoma, and gastrointestinal stromal tumors.
More innovative approaches include RNA-based therapies that inhibit the expression of VEGFR-2 or VEGF at the genetic level. Techniques like antisense oligonucleotides (ASOs), RNA interference (RNAi), and ribozyme-mediated degradation are being explored in preclinical and clinical settings to suppress aberrant angiogenesis from its origin.
In oncology, these therapies aim to “starve” tumors by cutting off their blood supply. In ophthalmology, particularly in age-related macular degeneration and diabetic retinopathy, anti-VEGFR-2 agents prevent vision loss by halting retinal neovascularization. Clinical trials are also exploring VEGFR-2 inhibition in inflammatory and neurodegenerative diseases, where vascular leakage and immune infiltration are driven by abnormal angiogenesis.
However, therapeutic targeting of VEGFR-2 is not without challenges. Resistance mechanisms, such as VEGF-independent angiogenesis or alternative pathway activation, can limit long-term efficacy. Therefore, combination therapies and biomarker-guided treatments are being actively pursued to optimize patient outcomes.
In conclusion, VEGFR-2 inhibition represents a powerful and versatile tool in modern medicine, with ongoing research poised to refine its use across a broader spectrum of diseases.
References
Carmeliet, P., Ferreira, V., Breier, G., Pollefeyt, S., Kieckens, L., Gertsenstein, M., … & Nagy, A. (1996). Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature, 380(6573), 435–439.
https://doi.org/10.1038/380435a0
Ferrara, N., & Kerbel, R. S. (2005). Angiogenesis as a therapeutic target. Nature, 438(7070), 967–974.
https://doi.org/10.1038/nature04483
Holmes, K., Roberts, O. L., Thomas, A. M., & Cross, M. J. (2007). Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signalling, 19(10), 2003–2012.
https://doi.org/10.1016/j.cellsig.2007.05.013
Shibuya, M. (2011). Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti-and pro-angiogenic therapies. Genes & Cancer, 2(12), 1097–1105.
https://doi.org/10.1177/1947601911423031
Croci, D. O., Cerliani, J. P., Dalotto-Moreno, T., Méndez-Huergo, S. P., Mascanfroni, I. D., Dergan-Dylon, S., … & Rabinovich, G. A. (2014). Glycosylation-dependent lectin–receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell, 156(4), 744–758.
https://doi.org/10.1016/j.cell.2014.01.043
Park, S. A., Jeong, M. S., Ha, K. T., & Jang, S. B. (2018). Structure and function of vascular endothelial growth factor and its receptor system. BMB Reports, 51(2), 73–78.
https://doi.org/10.5483/BMBRep.2018.51.2.233
Shibuya, M. (2013). VEGFR and type-V RTK activation and signaling. Cold Spring Harbor Perspectives in Biology, 5(7), a009092.
https://doi.org/10.1101/cshperspect.a009092
Takahashi, T., Yamaguchi, S., Chida, K., & Shibuya, M. (2001). A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-γ and DNA synthesis in vascular endothelial cells. EMBO Journal, 20(11), 2768–2778.
https://doi.org/10.1093/emboj/20.11.2768
Blanes, M. G., Oubaha, M., Rautureau, Y., & Gratton, J. P. (2007). Phosphorylation of tyrosine 801 of VEGFR-2 is necessary for Akt-dependent eNOS activation and NO release. Journal of Biological Chemistry, 282(15), 10660–10669.
https://doi.org/10.1074/jbc.M609048200
Holmqvist, K., Cross, M. J., Rolny, C., Hagerkvist, R., Rahimi, N., Matsumoto, T., et al. (2004). The adaptor protein SHB binds to tyrosine 1175 in VEGFR-2 and regulates VEGF-dependent migration. Journal of Biological Chemistry, 279(22), 22267–22275.
https://doi.org/10.1074/jbc.M312729200
Lamalice, L., Houle, F., & Huot, J. (2006). Phosphorylation of Tyr1214 within VEGFR-2 triggers the recruitment of Nck and activation of p38 MAPK pathway in endothelial cells. Journal of Biological Chemistry, 281(47), 34009–34020.
https://doi.org/10.1074/jbc.M603928200
Ferrara, N., & Adamis, A. P. (2016). Ten years of anti-vascular endothelial growth factor therapy. Nature Reviews Drug Discovery, 15(6), 385–403.
https://doi.org/10.1038/nrd.2015.17
Lugano, R., Ramachandran, M., & Dimberg, A. (2020). Tumor angiogenesis: causes, consequences, challenges and opportunities. Cellular and Molecular Life Sciences, 77(9), 1745–1770.
https://doi.org/10.1007/s00018-019-03351-7
Ma, Q. L., Ineichen, B. V., Detmar, M., & Proulx, S. T. (2017). Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nature Communications, 8, 1434.
https://doi.org/10.1038/s41467-017-01562-6
Yehya, A. H. S., Asif, M., Petersen, S. H., Subramaniam, A. V., Kono, K., Majid, A., et al. (2018). Angiogenesis: managing the culprits behind tumorigenesis and metastasis. Medicina, 54(1), 8.
https://doi.org/10.3390/medicina54010008
Fallah, A., Sadeghinia, A., Kahroba, H., Samadi, A., Heidari, H. R., Bradaran, B., et al. (2019). Therapeutic targeting of angiogenesis molecular pathways in angiogenesis-dependent diseases. Biomedicine & Pharmacotherapy, 110, 775–785.
https://doi.org/10.1016/j.biopha.2018.12.022
Jain, R. K., Duda, D. G., Clark, J. W., & Loeffler, J. S. (2006). Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nature Clinical Practice Oncology, 3(1), 24–40.
https://doi.org/10.1038/ncponc0403
Weis, S. M., & Cheresh, D. A. (2011). Tumor angiogenesis: molecular pathways and therapeutic targets. Nature Medicine, 17(11), 1359–1370.