Unlocking the Power of Antibody–Drug Conjugates: Advances in Linker Chemistry for Smarter Cancer Therapies
Abstract
Antibody–drug conjugates (ADCs) represent one of the most promising advances in targeted cancer therapy, combining the precision of monoclonal antibodies with the potency of cytotoxic agents. Central to their success is the linker chemistry, which dictates stability in circulation, controlled drug release, and therapeutic efficacy. This blog explores the evolution of ADC linker strategies, from early lysine and cysteine conjugations to cutting-edge site-specific technologies. By highlighting innovations and future directions, it provides insight into how linker design is shaping the next generation of safer, more effective cancer treatments.
The Promise of ADCs as Targeted Cancer Therapies
The concept of a “magic bullet” in medicine was first envisioned by Paul Ehrlich more than a century ago. He imagined a treatment that could selectively target disease-causing cells while sparing healthy tissues, delivering maximum impact with minimal side effects. Today, that vision is being realized through antibody–drug conjugates (ADCs), a rapidly growing class of targeted cancer therapies.
ADCs are complex molecules that combine three essential components: a monoclonal antibody, a cytotoxic payload, and a chemical linker that joins them together. The antibody provides exquisite specificity by recognizing tumor-associated antigens, while the payload delivers powerful cell-killing activity once internalized. The linker ensures that the toxic drug is released only inside cancer cells, making the entire system more selective than traditional chemotherapy.
This elegant design has already produced notable clinical successes. Adcetris® (brentuximab vedotin), approved in 2011, demonstrated remarkable efficacy in relapsed Hodgkin lymphoma by targeting CD30. Two years later, Kadcyla® (trastuzumab emtansine) was approved for HER2-positive breast cancer, offering improved outcomes for patients resistant to earlier antibody-based therapies. These approvals validated ADCs as a therapeutic class and paved the way for dozens of clinical candidates now in development. As of 2015, more than 40 ADCs were in clinical trials, a number that continues to grow.
What makes ADCs particularly promising is their ability to bridge the best of both worlds: the precision of antibody-based therapies and the potency of small-molecule drugs. Traditional chemotherapy often fails because of its lack of selectivity, causing widespread toxicity and limiting dosing. By contrast, ADCs can deliver highly cytotoxic drugs at concentrations that would be intolerable systemically, but safe when guided by an antibody.
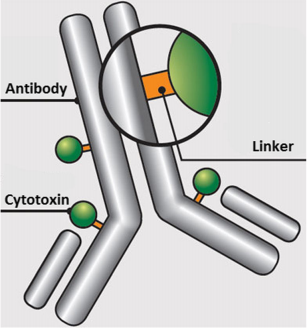
Fig. 1 Schematic of an ADC containing an antibody onto which is covalently attached a linker which in turn is covalently attached to a cytotoxin.
However, the road to success has not been straightforward. Early ADCs suffered from issues such as instability in the bloodstream, unpredictable pharmacokinetics, and off-target toxicities. The key lesson learned was that every component—antibody, payload, and particularly the linker chemistry—must be optimized for ADCs to achieve their full potential. The focus of current research is therefore not only on discovering new targets and payloads but also on designing smarter linkers that improve stability, enhance drug release, and reduce adverse effects.
As the field evolves, ADCs represent one of the most exciting frontiers in oncology. With continuous advances in linker chemistry and site-specific conjugation technologies, these “magic bullets” are moving closer to fulfilling Ehrlich’s century-old dream of highly effective and precise cancer therapy.
The Critical Role of Linkers in ADC Design
When discussing antibody–drug conjugates (ADCs), much attention is often given to the antibody’s ability to target cancer cells or the potency of the cytotoxic payload. Yet, the linker—the chemical bridge that holds these two elements together—plays an equally vital role in determining whether an ADC succeeds or fails.
The ideal linker must balance two seemingly contradictory properties: stability in the bloodstream and efficient release inside the tumor cell. Stability is essential because premature release of a highly toxic drug can cause serious off-target damage and limit the therapeutic index. At the same time, once the ADC is internalized into the cancer cell, the linker must reliably release the payload to ensure tumor cell death. Achieving this balance has been one of the greatest challenges in ADC design.
Linkers are broadly classified into cleavable and non-cleavable types. Cleavable linkers are designed to exploit specific intracellular conditions. For example, peptide-based linkers such as valine-citrulline (vc) are sensitive to proteases like cathepsin B, which are abundant in tumor lysosomes. Acid-sensitive linkers, such as hydrazones, take advantage of the lower pH inside endosomes and lysosomes compared to blood plasma. Other linkers, such as disulfides, exploit the higher intracellular concentration of glutathione to trigger release. These strategies ensure that the toxic payload is liberated selectively within cancer cells.
In contrast, non-cleavable linkers rely on the complete degradation of the antibody within the lysosome. The payload remains attached to an amino acid or linker fragment, which can still be cytotoxic. An example is the MCC-DM1 construct used in Kadcyla® (trastuzumab emtansine), where the stability of the linker results in a longer half-life and reduced off-target toxicity.
Importantly, linkers also influence other properties of ADCs beyond release. Hydrophobic linkers, for instance, can increase the tendency of ADCs to aggregate, potentially leading to hepatotoxicity and immunogenicity. Conversely, more hydrophilic linkers improve solubility and may reduce recognition by multidrug resistance transporters, enhancing therapeutic effectiveness.
Ultimately, the design of an ADC is not simply about attaching a drug to an antibody. The linker is the control switch that dictates where, when, and how the toxic payload is unleashed. Optimizing linker chemistry is therefore at the heart of ADC innovation, ensuring these therapies achieve maximum precision with minimal collateral damage.
Current Strategies: Cysteine vs. Lysine Conjugation
A central challenge in antibody–drug conjugate (ADC) development is how to attach the cytotoxic drug to the antibody in a way that is both stable and reproducible. The two dominant strategies in clinical use today involve cysteine conjugation and lysine conjugation. Each approach has distinct advantages and limitations, influencing drug–antibody ratio (DAR), homogeneity, and ultimately clinical performance.
Lysine conjugation was the first widely adopted method. Lysines are abundant on antibodies—an IgG1, for example, has roughly 90 lysine residues, with about 30 available for modification. In this strategy, a linker containing a reactive group attaches to lysine side chains, typically through acylation. While straightforward, this approach creates a highly heterogeneous mixture, since the drug can attach to many different sites on the antibody surface. The resulting ADC population may have variable DARs, often averaging between three and four. Such heterogeneity can affect antigen binding, pharmacokinetics, and stability. A well-known example is Kadcyla® (trastuzumab emtansine), which uses an MCC-DM1 linker–drug system attached via lysine residues. Despite its heterogeneity, Kadcyla’s clinical success demonstrates the viability of this approach.
Cysteine conjugation, by contrast, relies on reducing the antibody’s interchain disulfide bonds to expose free thiol groups for reaction with maleimide-based linkers. Since an IgG1 contains only four interchain disulfides, there are at most eight available conjugation sites, resulting in lower heterogeneity compared to lysine methods. This approach typically yields ADCs with an even-numbered DAR, most often four. The strategy has been instrumental in the development of Adcetris® (brentuximab vedotin), which uses a cysteine–maleimide linkage with a protease-cleavable valine-citrulline (vc) linker.
Both methods have trade-offs. Lysine conjugation preserves the antibody’s disulfide architecture but introduces significant heterogeneity. Cysteine conjugation produces more uniform ADCs but can weaken antibody structural stability due to disulfide reduction. To address these limitations, researchers are exploring engineered cysteine sites and site-specific conjugation strategies that improve homogeneity and therapeutic index.
In short, lysine and cysteine conjugation represent the first generation of ADC attachment strategies. Their continued refinement—and eventual transition to site-specific methods—will define how future ADCs achieve greater safety, consistency, and efficacy.
Innovations in Site-Specific Conjugation
While traditional lysine and cysteine conjugation strategies have enabled the clinical success of ADCs, their inherent heterogeneity remains a major limitation. Variability in the number and location of attached drug molecules can affect pharmacokinetics, potency, and safety. To overcome this challenge, researchers have turned to site-specific conjugation technologies, which allow for precise attachment of cytotoxic payloads at defined locations on the antibody. This innovation represents a major step forward in the design of next-generation ADCs.
One widely used approach involves engineered cysteines. By introducing additional cysteine residues at predetermined positions in the antibody structure, researchers can control where drug molecules are attached. This method, pioneered in the development of THIOMABs, significantly reduces heterogeneity while maintaining antigen-binding activity. Site-specific cysteine conjugation has shown improved tolerability and therapeutic index in preclinical models compared to conventional approaches.
Another powerful strategy employs non-natural amino acids (nnAAs) engineered into the antibody sequence. For example, p-acetylphenylalanine (pAcPhe) introduces a carbonyl group that can react with linker–drug molecules via oxime chemistry. Similarly, azido-containing nnAAs enable conjugation through copper-free click chemistry, such as strain-promoted azide–alkyne cycloaddition (SPAAC). These methods open the door to highly selective bioconjugation while expanding the chemical diversity of linkers and payloads that can be used.
Enzyme-mediated conjugation offers another exciting frontier. Enzymes such as formylglycine-generating enzyme (FGE) can introduce aldehyde groups into antibodies, which then serve as unique handles for further conjugation. This technology, known as SMARTag, enables stable, site-specific modification without disrupting antibody function. Other enzymes, like bacterial transglutaminase or sortase A, can recognize short peptide motifs engineered into antibodies and catalyze covalent attachment of drugs at precise sites.
The advantages of site-specific approaches are clear: improved homogeneity, consistent drug–antibody ratios, enhanced stability, and often a wider therapeutic window. These technologies not only refine current ADC platforms but also provide opportunities to incorporate novel bio-orthogonal chemistries, broadening the range of possible linker and payload designs.
As site-specific methods advance toward clinical application, they are expected to play a central role in shaping the future of ADCs. By ensuring precision at the molecular level, these technologies bring us closer to realizing the full therapeutic potential of antibody–drug conjugates in oncology and beyond.
Future Directions: Toward Smarter, Safer ADCs
The field of antibody–drug conjugates (ADCs) has evolved rapidly over the past two decades, yet challenges remain in achieving maximum precision, safety, and therapeutic benefit. As next-generation ADCs emerge, researchers are focusing on refining linker chemistry, drug payloads, and site-specific conjugation strategies to overcome limitations seen in earlier designs.
One key challenge is aggregation and stability. Many cytotoxic payloads are highly hydrophobic, and when linked to antibodies, they can promote aggregation. Aggregates not only reduce drug solubility but may also increase immunogenicity and hepatotoxicity. Future efforts are directed at developing hydrophilic linkers, such as polyethylene glycol (PEG)-based or sulfonated spacers, which improve solubility and reduce aggregation risks. Such modifications may also help ADCs bypass multidrug resistance (MDR) transporters, thereby enhancing intracellular drug retention.
Another critical direction involves bio-orthogonal chemistries. Traditional cysteine and lysine conjugation strategies, while effective, introduce heterogeneity. Site-specific conjugation with engineered cysteines, non-natural amino acids, or enzymatic handles allows for homogeneous ADC populations with well-defined drug–antibody ratios (DARs). These precise technologies not only improve therapeutic consistency but also enable the use of novel linkers that respond to unique intracellular conditions, further enhancing selective drug release.
Researchers are also exploring novel payload classes. While maytansinoids and auristatins have dominated clinical ADC development, emerging payloads include DNA alkylators, topoisomerase inhibitors, and immune modulators. These expand the therapeutic reach of ADCs beyond traditional tubulin-targeting agents and may provide solutions for tumors resistant to first-generation ADCs.
Importantly, the future of ADCs will also be shaped by their integration into combination therapies. ADCs paired with checkpoint inhibitors, kinase inhibitors, or immunotherapies could synergistically enhance antitumor activity while reducing resistance. This integrated approach reflects a broader trend toward personalized medicine, where ADCs are tailored to tumor-specific markers and combined with complementary modalities for maximum effect.
In summary, the next wave of ADC development is poised to deliver smarter, safer, and more effective cancer therapies. By combining advances in linker chemistry, site-specific engineering, and novel payload discovery, researchers are steadily transforming Paul Ehrlich’s century-old “magic bullet” concept into a powerful clinical reality. The ongoing evolution of ADCs promises not just incremental improvements but a new era in precision oncology.
References
Jain, N., Smith, S. W., Ghone, S., & Tomczuk, B. (2015). Current ADC linker chemistry. Pharmaceutical Research, 32(11), 3526–3540.
https://doi.org/10.1007/s11095-015-1657-7
Senter, P. D., & Sievers, E. L. (2012). The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nature Biotechnology, 30(7), 631–637.
https://doi.org/10.1038/nbt.2289
Phillips, G. D. L., Li, G., Dugger, D. L., Crocker, L. M., Parsons, K. L., Mai, E., & et al. (2008). Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody–drug conjugate. Cancer Research, 68(22), 9280–9290.
https://doi.org/10.1158/0008-5472.CAN-08-1776
Trail, P. A. (2013). Antibody drug conjugates as cancer therapeutics. Antibodies, 2(1), 113–129.
https://doi.org/10.3390/antib2010113
Dubowchik, G. M., & Firestone, R. A. (1998). Cathepsin B-sensitive dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjugate Chemistry, 9(4), 500–511.
https://doi.org/10.1021/bc980037s
Teicher, B. A., & Chari, R. V. J. (2011). Antibody conjugate therapeutics: Challenges and potential. Clinical Cancer Research, 17(20), 6389–6397.
https://doi.org/10.1158/1078-0432.CCR-11-1427
Jununtula, J. R., Raab, H., Clark, S., Bhakta, S., Leipold, D. D., Weir, S., … & Scheller, R. H. (2008). Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nature Biotechnology, 26(8), 925–932.