Tyrosine Kinases in Cancer: From Molecular Mechanisms to Targeted Therapies
Abstract
Tyrosine kinases play a pivotal role in cellular signaling pathways that regulate proliferation, differentiation, metabolism, and apoptosis. In cancer, mutations, gene fusions, and autocrine signaling loops can lead to the aberrant activation of these kinases, resulting in unchecked tumor growth and survival. This blog explores the mechanisms behind tyrosine kinase–driven oncogenesis and highlights both established and emerging therapeutic strategies targeting these enzymes. From first-generation inhibitors like Imatinib and monoclonal antibodies such as Herceptin to innovative approaches involving Hsp90 inhibition, immunotoxins, and antisense technologies, the landscape of targeted therapy is rapidly evolving. The article also looks ahead to the future of personalized cancer care through kinome profiling, structure-based drug design, and functional genomics. By understanding the role of tyrosine kinases in cancer, researchers and clinicians are paving the way for more precise, effective, and individualized treatments.
Introduction to Tyrosine Kinases and Their Role in Cancer
Tyrosine kinases (TKs) are enzymes that act as essential regulators in cellular signaling networks, transmitting extracellular signals into precise intracellular actions. These proteins catalyze the transfer of phosphate groups from ATP to tyrosine residues on specific substrate proteins—a post-translational modification that initiates or modulates critical cellular processes such as proliferation, differentiation, metabolism, and programmed cell death.
In healthy cells, the activity of tyrosine kinases is tightly controlled. They are activated only in response to specific stimuli, such as the binding of growth factors to cell surface receptors. However, when this regulation breaks down, tyrosine kinases can become perpetually active, driving uncontrolled cell growth and contributing to the transformation of normal cells into malignant ones.
Deregulation of tyrosine kinases is one of the hallmarks of many human cancers. Mutations, gene amplifications, and chromosomal translocations can lead to the constant activation of these enzymes. For instance, the BCR-ABL fusion gene, resulting from a chromosomal translocation, creates a constitutively active tyrosine kinase that is a defining feature of chronic myeloid leukemia (CML). Similarly, mutations in the EGFR gene can promote tyrosine kinase activation in non-small cell lung cancer and glioblastoma.
With more than 90 tyrosine kinases identified in the human genome, their centrality in cancer biology has made them prominent targets in precision oncology. By disrupting abnormal TK signaling, researchers and clinicians aim to halt tumor progression at the molecular level.
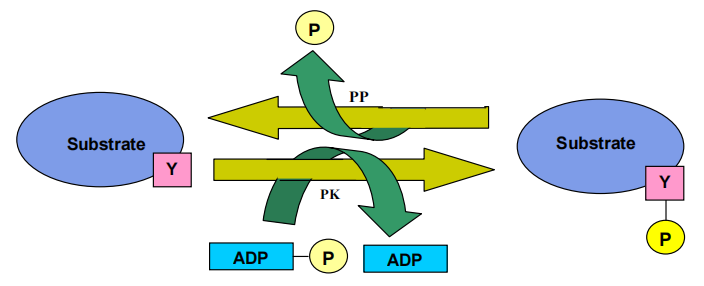
Figure 1. Schematic representation of the mode of action of tyrosine kinase. PK represents protein kinase and PP stands for protein phosphatase.
Understanding the role of tyrosine kinases in cancer is fundamental to developing targeted therapies that offer higher specificity and fewer side effects compared to conventional chemotherapy. As research advances, these molecular gatekeepers are not just markers of disease but also gateways to personalized, effective cancer treatments.
Mechanisms Behind Oncogenic Activation of Tyrosine Kinases
Under normal physiological conditions, tyrosine kinase activity is carefully regulated through feedback loops and phosphatase activity. However, in many cancers, this balance is disrupted, leading to constitutive activation of tyrosine kinases and persistent downstream signaling that promotes malignant transformation. There are three primary mechanisms behind this deregulation: genetic mutations, chromosomal translocations, and autocrine or paracrine stimulation loops.
Genetic Mutations
Point mutations or deletions in tyrosine kinase genes can render the kinase constitutively active, bypassing the need for ligand binding. A well-documented example is the EGFRvIII mutation, which deletes amino acids in the extracellular domain of the epidermal growth factor receptor (EGFR), resulting in ligand-independent activation. This mutation has been identified in glioblastoma multiforme, non-small cell lung cancer (NSCLC), and ovarian tumors.
Chromosomal Translocations
Gene fusions caused by chromosomal rearrangements are another major driver of tyrosine kinase activation. In chronic myeloid leukemia (CML), a reciprocal translocation between chromosomes 9 and 22 creates the BCR-ABL fusion gene. The resulting fusion protein has significantly enhanced tyrosine kinase activity and promotes unchecked cell proliferation. Similar fusions such as TEL-ABL and TEL-PDGFR have been observed in various forms of leukemia.
Autocrine and Paracrine Stimulation Loops
Cancers can also sustain tyrosine kinase activation through overexpression of both receptors and their ligands. In an autocrine loop, a cancer cell produces both the growth factor and its receptor, leading to continuous stimulation. For instance, EGFR and its ligands EGF and TGF-α are frequently co-expressed in lung, bladder, and breast cancers. Similar loops involving PDGFR and IGF-1R contribute to tumor progression in gliomas and prostate cancer, respectively.
These mechanisms enable cancer cells to evade normal regulatory cues, avoid apoptosis, and sustain proliferative signaling—hallmarks of malignancy.
Targeting Tyrosine Kinases in Modern Cancer Therapy
The discovery of the pivotal role tyrosine kinases play in cancer progression has transformed the therapeutic landscape, ushering in an era of targeted therapies that disrupt oncogenic signaling at its source. Rather than relying solely on traditional chemotherapeutics that indiscriminately affect rapidly dividing cells, modern approaches focus on inhibiting specific tyrosine kinases that drive tumor growth and survival.
Small Molecule Inhibitors
One of the most notable breakthroughs came with the development of Imatinib mesylate (Gleevec), a small molecule inhibitor targeting the BCR-ABL fusion protein in chronic myeloid leukemia (CML). Gleevec functions by occupying the ATP-binding site of the ABL kinase, thereby preventing phosphorylation and halting downstream proliferative signals. Its success not only validated tyrosine kinases as drug targets but also demonstrated the power of precision medicine in oncology.
Following Gleevec’s success, other ATP-competitive inhibitors emerged. Gefitinib (Iressa) and Erlotinib (Tarceva) were developed to target mutant forms of EGFR, commonly seen in non-small cell lung cancer (NSCLC). These drugs have shown efficacy in subsets of patients harboring activating EGFR mutations, offering a more personalized treatment strategy.
Monoclonal Antibodies
While small molecules target the intracellular kinase domain, monoclonal antibodies like Trastuzumab (Herceptin) bind to extracellular regions of receptor tyrosine kinases. Herceptin specifically targets HER2, a member of the EGFR family, and is used in HER2-positive breast cancers. By binding to HER2, it not only inhibits downstream signaling but also flags the cancer cell for immune-mediated destruction.
These targeted therapies highlight a paradigm shift in cancer treatment—from broad-spectrum cytotoxic agents to molecularly guided interventions. However, resistance often develops, necessitating combination strategies and the development of next-generation inhibitors. Still, targeting tyrosine kinases remains one of the most successful and actively pursued strategies in contemporary oncology.
Emerging Strategies in Tyrosine Kinase Inhibition
As resistance to first-generation tyrosine kinase inhibitors (TKIs) and monoclonal antibodies emerges, researchers have turned to innovative strategies to expand the arsenal against tyrosine kinase-driven cancers. These next-generation approaches aim to overcome drug resistance, enhance specificity, and exploit alternative biological vulnerabilities.
Hsp90 Inhibitors
Many oncogenic kinases rely on molecular chaperones like Heat Shock Protein 90 (Hsp90) to maintain their functional conformations. Inhibitors such as Geldanamycin and its derivative 17-AAG bind to the ATP-binding pocket of Hsp90, disrupting its stabilizing effect and promoting degradation of client oncoproteins like HER2, EGFR, and BCR-ABL. By targeting these support systems, Hsp90 inhibitors offer a powerful way to shut down multiple cancer pathways simultaneously.
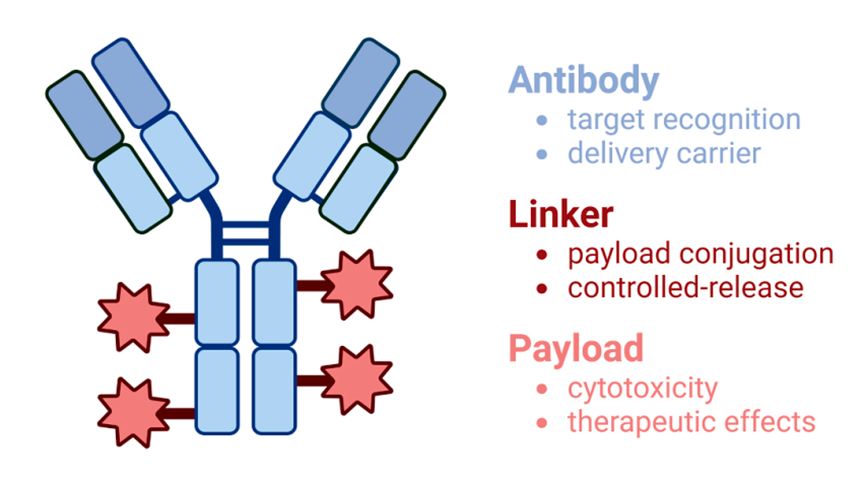
Immunotoxins and Antibody-Drug Conjugates (ADCs)
Another promising approach involves combining monoclonal antibodies with cytotoxic agents to form immunotoxins or antibody-drug conjugates. These hybrid molecules selectively bind cancer-specific receptors (e.g., EGFR, CD25) and deliver toxins directly into malignant cells. For instance, DAB389EGF, a conjugate of EGF and diphtheria toxin, has shown efficacy in EGFR-overexpressing tumors, minimizing harm to normal tissues.
Antisense Oligonucleotides and Peptidomimetics
Antisense oligonucleotides (ASOs) inhibit tyrosine kinase expression by binding to mRNA transcripts, preventing translation. Clinical candidates like ISIS 3521, which targets PKC-α, have demonstrated potential in non-small cell lung cancer and breast cancer. Meanwhile, peptidomimetics block critical protein–protein interactions in kinase signaling cascades, such as the Grb2-Sos interaction in the Ras pathway.
Anti-Angiogenic TK Inhibitors
Because tyrosine kinases like VEGFR and FGFR drive angiogenesis, targeting them with inhibitors like SU5416 and PD173074 can deprive tumors of blood supply. Unlike conventional drugs, anti-angiogenic therapies show lower rates of acquired resistance, making them attractive for long-term treatment strategies.
Together, these emerging approaches reflect a broader trend: turning the complexity of cancer signaling into therapeutic opportunity, with tyrosine kinases remaining a critical focal point.
Future Perspectives – Mining the Kinome for Personalized Oncology
The future of cancer therapy lies in personalized, molecularly targeted interventions, and tyrosine kinases continue to occupy a central role in this shift. With over 500 kinases encoded in the human genome, the concept of the “kinome”—the full complement of protein kinases—offers an expansive landscape for therapeutic exploration. Many cancers are “oncogene-addicted,” relying heavily on a single aberrant signaling pathway for growth and survival. This vulnerability presents an opportunity: by identifying and selectively targeting these kinases, clinicians can disrupt cancer progression with minimal harm to normal tissues.
However, mining the kinome is not without challenges. The high degree of homology among kinase domains, particularly in ATP-binding regions, necessitates the development of highly selective inhibitors to avoid off-target effects. Moreover, tumors often adapt through compensatory pathways or mutations, leading to resistance. As a result, modern drug development must go beyond simple inhibitor screening.
Innovative strategies are being adopted to accelerate tyrosine kinase drug discovery. High-throughput screening, computational modeling, and structure-based drug design (SBDD) are now essential tools. These approaches allow researchers to predict binding affinities, evaluate selectivity, and optimize lead compounds more efficiently than traditional trial-and-error methods.
Equally promising is the application of RNA interference (RNAi) and CRISPR-based genetic editing to functionally validate kinase targets. These technologies help identify which kinases are essential for tumor survival, offering a more rational approach to drug development. Furthermore, advances in cancer genomics and proteomics now enable clinicians to profile tumors at the molecular level, tailoring treatments to each patient’s unique kinase expression and mutation profile.
In the era of precision oncology, the integration of kinome-wide screening, genomic diagnostics, and adaptive therapeutic strategies promises to transform how we diagnose, monitor, and treat cancer. The goal is clear: to deliver targeted, durable, and personalized therapies that strike cancer at its molecular roots.
References
Blume-Jensen, P., & Hunter, T. (2001). Oncogenic kinase signalling. Nature, 411(6835), 355–365.
https://doi.org/10.1038/35077225
Hunter, T. (2000). Signaling—2000 and beyond. Cell, 100(1), 113–127.
https://doi.org/10.1016/S0092-8674(00)81688-8
Schlessinger, J. (2000). Cell signaling by receptor tyrosine kinases. Cell, 103(2), 211–225.
https://doi.org/10.1016/S0092-8674(00)00114-8
Andrews, D. W., Resnicoff, M., Flanders, A. E., Kenyon, L., Curtis, M., Merli, G., Baserga, R., Iliakis, G., & Aiken, R. D. (2001). A pilot study of antisense therapy targeting insulin-like growth factor type I receptor in malignant astrocytomas. Journal of Clinical Oncology, 19(8), 2189–2200.
https://doi.org/10.1200/JCO.2001.19.8.2189
Daley, G. Q., Van Etten, R. A., & Baltimore, D. (1990). Induction of chronic myelogenous leukemia in mice by the P210 BCR-ABL gene. Science, 247(4944), 824–830.
https://doi.org/10.1126/science.2406902
Druker, B. J., Talpaz, M., Resta, D. J., Peng, B., Buchdunger, E., Ford, J. M., … & Sawyers, C. L. (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. New England Journal of Medicine, 344(14), 1031–1037.
https://doi.org/10.1056/NEJM200104053441401
Dunn, D. A. (2002). Mining the human ‘kinome’. Drug Discovery Today, 7(22), 1121–1123.
https://doi.org/10.1016/S1359-6446(02)02588-1
Fukuoka, M., Yano, S., Giaccone, G., Tamura, T., Nakagawa, K., Douillard, J. Y., … & Nishiwaki, Y. (2003). Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non–small-cell lung cancer. Journal of Clinical Oncology, 21(12), 2237–2246.
https://doi.org/10.1200/JCO.2003.10.038
Kreitman, R. J. (1999). Immunotoxins in cancer therapy. Current Opinion in Immunology, 11(5), 570–578.
https://doi.org/10.1016/S0952-7915(99)00012-5
Nishikawa, R. E. A., Ji, X. D., Harmon, R. C., Lazar, C. S., Gill, G. N., Cavenee, W. K., & Huang, H. J. S. (1994). A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proceedings of the National Academy of Sciences, 91(16), 7727–7731.
https://doi.org/10.1073/pnas.91.16.7727
Paul, M. K., & Mukhopadhyay, A. K. (2004). Tyrosine kinase–Role and significance in cancer. International Journal of Medical Sciences, 1(2), 101–115.
https://doi.org/10.7150/ijms.1.101
Slamon, D. J., Leyland-Jones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A., … & Norton, L. (2001). Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. New England Journal of Medicine, 344(11), 783–792.
https://doi.org/10.1056/NEJM200103153441101
Sreedhar, A. S., Soti, C., & Csermely, P. (2003). Inhibition of Hsp90: A new strategy for inhibiting protein kinases. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research, 1603(2), 122–135.
https://doi.org/10.1016/S0304-4165(02)00318-0
Workman, P. (2001). Scoring a bull’s-eye against cancer genome targets. Current Opinion in Pharmacology, 1(4), 342–352.
https://doi.org/10.1016/S1471-4892(01)00081-2
Zwick, E., Bange, J., & Ullrich, A. (2002). Receptor tyrosine kinases as targets for anticancer drugs. Trends in Molecular Medicine, 8(1), 17–23.