The PI3K Pathway in Disease: From Cancer Mutations to Precision Therapy and Immune Modulation
Abstract
The phosphoinositide 3-kinase (PI3K) signaling pathway plays a pivotal role in regulating cellular processes such as growth, metabolism, survival, and immune response. This blog post explores the evolving understanding of PI3K in human disease, focusing on its molecular structure, disease-associated mutations, and clinical relevance. We highlight how PI3K dysregulation contributes to cancer, immune disorders, and metabolic dysfunction, while also addressing the challenges and opportunities of targeting this pathway therapeutically. Special attention is given to current drug development strategies, resistance mechanisms, and the integration of PI3K inhibitors into precision medicine and immunotherapy. With new tools and refined approaches, the PI3K pathway stands at the crossroads of translational research and clinical innovation.
Decoding the PI3K Pathway: Why It Matters in Health and Disease
In the complex circuitry of cellular communication, few signaling pathways are as pivotal as the phosphoinositide 3-kinase (PI3K) pathway. This highly conserved molecular network governs essential cellular functions such as growth, metabolism, survival, proliferation, and immune response. As research over the past three decades has shown, dysregulation of PI3K signaling is a major driver of diverse human diseases—from cancer to immune deficiencies and metabolic disorders.
Originally identified in the 1980s through studies of oncogene-associated lipid phosphorylation, PI3K emerged as a central mediator of signals downstream of growth factor receptors and oncogenic proteins. The key bioactive lipid produced by PI3K enzymes, PtdIns(3,4,5)P₃, acts as a second messenger, recruiting a suite of proteins to the plasma membrane and triggering cascades that ultimately regulate cell fate.
In cancer, aberrant PI3K activation has been linked to uncontrolled cell proliferation, resistance to cell death, and metabolic reprogramming—traits that make this pathway a hallmark of tumor biology. Mutations in the PIK3CA gene, which encodes the p110α catalytic subunit of class I PI3K, are among the most common alterations across human tumors, especially in breast, colon, and endometrial cancers. Similarly, the tumor suppressor PTEN, which antagonizes PI3K signaling, is frequently deleted or mutated in malignancies.
Beyond oncology, PI3K has gained recognition for its role in immune system regulation. Isoform-specific mutations are known to cause primary immunodeficiencies, such as activated PI3K-δ syndrome (APDS), while the same pathway influences immune cell activation and differentiation. Moreover, PI3K signaling is deeply intertwined with insulin action and glucose homeostasis, making it a focal point in the study of obesity, insulin resistance, and type 2 diabetes.
As this review series explores, understanding the nuanced roles of PI3K signaling is critical—not only for targeting disease with precision but also for avoiding systemic side effects that arise from disrupting a pathway so deeply embedded in physiology.
The Core Mechanics of PI3K Signaling: How Cellular Signals Become Action
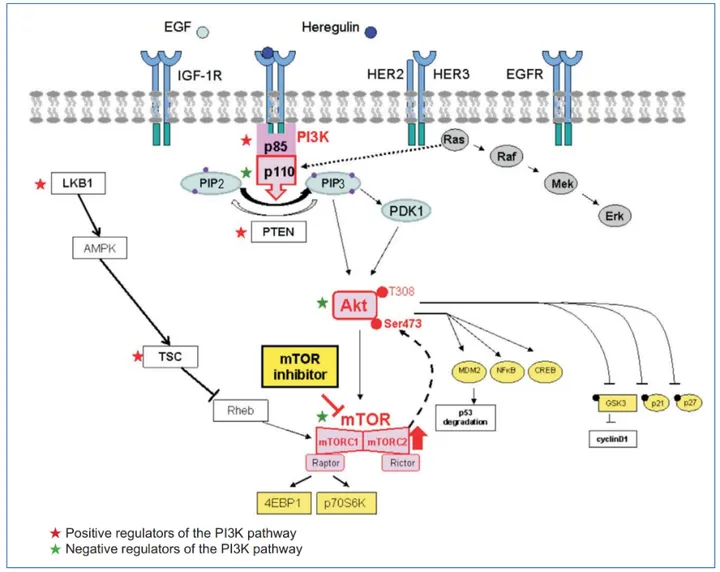
At the heart of many cellular decisions—whether a cell grows, divides, survives, or migrates—is the phosphoinositide 3-kinase (PI3K) pathway. This signaling cascade begins with class I PI3Ks, lipid kinases that are activated by diverse extracellular stimuli such as growth factors, cytokines, and chemokines. Once activated, these enzymes generate the lipid messenger phosphatidylinositol (3,4,5)-trisphosphate (PIP₃), which orchestrates a broad array of downstream signaling events.
Class I PI3Ks are composed of four catalytic isoforms: p110α, p110β, p110γ, and p110δ. These are encoded by the genes PIK3CA, PIK3CB, PIK3CG, and PIK3CD, respectively. The p110α and p110β isoforms are expressed ubiquitously, whereas p110γ and p110δ are predominantly found in leukocytes, reflecting their specialized roles in the immune system.
Each catalytic subunit forms a dimer with a regulatory subunit that modulates its activity and localization. Class IA PI3Ks (p110α, β, and δ) are typically activated downstream of receptor tyrosine kinases (RTKs) through SH2-domain-containing regulatory subunits (like p85), which bind phosphorylated tyrosine residues on activated receptors. In contrast, class IB PI3K (p110γ) is activated downstream of G protein-coupled receptors (GPCRs) via interaction with Gβγ subunits and its regulatory proteins p101 or p87.
Once generated, PIP₃ recruits proteins with pleckstrin homology (PH) domains—most notably AKT (also known as protein kinase B) and PDK1—to the plasma membrane. This initiates a phosphorylation cascade that culminates in AKT activation, which regulates numerous substrates involved in glucose metabolism, cell survival, and proliferation.
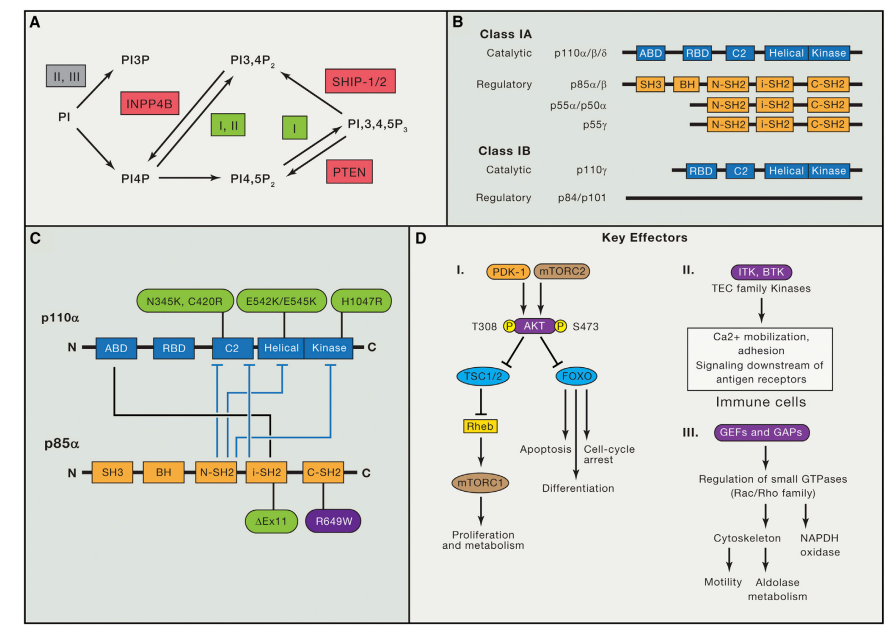
Figure 1. Overview of Phosphoinositides, Class I PI3K Protein Isoforms, p110a Activity Regulation, and PI3K Downstream Effectors.
The PI3K pathway also interfaces with the mechanistic target of rapamycin (mTOR), forming a critical axis for coordinating nutrient sensing, energy balance, and biosynthetic processes. The intricate interplay among PI3K isoforms, their activators, and effectors allows cells to tailor their responses precisely to environmental cues. However, this complexity also presents challenges—disruptions in this balance underlie many diseases, particularly cancer and immune disorders.
Understanding the structural and biochemical logic of PI3K activation is essential for designing targeted therapies that exploit its vulnerabilities while preserving essential cellular functions.
PI3K Mutations and Their Role in Human Disease
Mutations in the PI3K signaling pathway have profound consequences for human health. While originally identified for its role in cancer, recent discoveries have expanded our understanding of how mutations in PI3K genes contribute to a broader spectrum of diseases, including immunodeficiencies and developmental disorders.
The gene PIK3CA, encoding the p110α catalytic subunit of class I PI3K, is among the most frequently mutated oncogenes in human cancer. Mutations such as H1047R and E545K result in constitutive activation of the enzyme, bypassing the need for upstream growth factor signaling and promoting persistent proliferative and survival signals in cancer cells. These alterations are particularly prevalent in breast, colorectal, and endometrial cancers, and often coexist with other mutations that drive tumor progression.
Loss-of-function mutations in the tumor suppressor PTEN, which counteracts PI3K by dephosphorylating PIP₃, are also common across cancers. The combined effect of increased PI3K activity and impaired negative regulation leads to sustained AKT and mTOR signaling—hallmarks of aggressive tumor biology.
Beyond oncology, germline or somatic mutations in PI3K pathway components cause a range of non-malignant diseases. For instance, somatic PIK3CA mutations give rise to mosaic overgrowth syndromes such as CLOVES (congenital lipomatous overgrowth with vascular, epidermal, and skeletal anomalies). These disorders feature localized tissue hyperplasia due to the same mutations that drive cancer, but arising during development.
In the immune system, activating mutations in PIK3CD (encoding p110δ) lead to Activated PI3K-Delta Syndrome (APDS), an inborn error of immunity characterized by recurrent infections, lymphoproliferation, and autoimmunity. These mutations elevate PI3K signaling in lymphocytes, causing functional exhaustion and senescence. Similarly, mutations in the regulatory subunit gene PIK3R1 result in APDS2, which shares overlapping clinical features.
These examples underscore the PI3K pathway’s critical balance—too much or too little activity can disrupt normal development, immune surveillance, and cellular homeostasis. Therapeutic strategies now seek to modulate PI3K signaling precisely, with isoform-selective inhibitors and novel approaches to avoid collateral damage to healthy tissues.
Targeting the PI3K Pathway: Opportunities and Therapeutic Challenges
The PI3K signaling cascade has become a prime target in precision oncology, given its frequent dysregulation in cancers. Inhibitors of this pathway—particularly those targeting class I PI3K isoforms, AKT, or mTOR—have shown promise in both hematological malignancies and solid tumors. Yet despite decades of preclinical and clinical effort, therapeutic success has been mixed, revealing both the potential and complexity of targeting such a central signaling hub.
One of the key breakthroughs was the FDA approval of isoform-selective PI3K inhibitors, such as idelalisib, a p110δ inhibitor, for chronic lymphocytic leukemia and follicular lymphoma. Its effectiveness in B-cell malignancies stems from the critical role p110δ plays in B-cell receptor signaling. Meanwhile, alpelisib, a p110α-specific inhibitor, has shown efficacy in PIK3CA-mutated breast cancers, especially when combined with endocrine therapy.
However, these advances have been tempered by several challenges. First, on-target toxicities remain a major issue. Because PI3K enzymes regulate normal glucose metabolism, inhibition—particularly of p110α—often leads to hyperglycemia, insulin resistance, and compensatory insulin release, which may blunt drug efficacy in tumors.
Second, intrinsic and acquired resistance severely limit the durability of response. Tumors often escape PI3K inhibition through feedback loops (e.g., reactivation of AKT or ERK signaling), isoform switching (from p110α to p110β), or activation of parallel pathways like JAK/STAT or MYC. These compensatory mechanisms restore survival signals despite PI3K blockade, necessitating rational combination strategies.
To address these issues, researchers are exploring combination therapies—pairing PI3K inhibitors with CDK4/6 inhibitors, anti-estrogens, MEK inhibitors, or even immune checkpoint therapies. High-dose intermittent dosing regimens are also being evaluated to maximize tumor cell kill while allowing normal tissues to recover, potentially improving tolerability without sacrificing efficacy.
Ultimately, while the PI3K pathway remains a valuable target in oncology, its pleiotropic roles in normal physiology demand precision in both patient selection and therapeutic design. Future success hinges on understanding resistance biology, minimizing systemic toxicity, and tailoring therapy based on tumor genotype and context.
PI3K at the Crossroads of Precision Medicine: Future Directions
As our understanding of the phosphoinositide 3-kinase (PI3K) pathway deepens, so too does its potential to reshape precision medicine. No longer viewed solely as a driver of cancer cell proliferation, PI3K signaling is now recognized as a crucial interface between metabolism, immunity, and tumor microenvironment regulation. This broader perspective is redefining how researchers and clinicians approach therapy design and patient stratification.
One of the most promising frontiers lies in combining PI3K-targeted agents with immunotherapy. Recent studies have shown that tumor-intrinsic PI3K activation—especially through PTEN loss—can promote immune evasion by suppressing T-cell infiltration and enhancing PD-L1 expression. In preclinical models, selective inhibition of p110β or mTORC1 has restored antitumor immunity and enhanced the efficacy of anti-PD-1 therapy. These findings suggest that the PI3K pathway not only fuels tumor growth but also shapes the immune landscape—making it an ideal co-target in cancer immunotherapy.
Another emerging focus is isoform-selective drug development and mutant-specific inhibitors. For example, next-generation compounds aim to selectively inhibit oncogenic PIK3CA mutations (such as H1047R) while sparing wild-type isoforms that are critical for normal metabolism. This approach could significantly reduce systemic toxicity, especially in insulin-sensitive tissues, and allow for longer-term administration.
Additionally, temporal and spatial dynamics of PI3K activity are gaining attention. Tumors are often genetically heterogeneous, and subclonal PI3K pathway activation can vary across primary and metastatic sites. Precision therapies will likely need to account for these differences through advanced molecular profiling, potentially using circulating tumor DNA (ctDNA) to monitor resistance and guide treatment decisions in real time.
Finally, alternative dosing strategies—such as intermittent high-dose regimens—are being investigated to overcome feedback resistance and reduce toxicity. These strategies exploit the differential recovery rates of tumor versus normal tissue and may offer a better therapeutic window than continuous dosing.
In the coming years, progress in PI3K-targeted therapy will depend not only on drug innovation but also on integrative approaches that combine genomics, immunology, and metabolic profiling. The pathway’s central role in diverse biological processes positions it as a linchpin in the next generation of personalized cancer treatment.
References
Lucas, C. L., Kuehn, H. S., Zhao, F., Niemela, J. E., Deenick, E. K., Palendira, U., … & Lenardo, M. J. (2016). Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nature Immunology, 15(1), 88–97.
https://doi.org/10.1038/ni.2771
Samuels, Y., Wang, Z., Bardelli, A., Silliman, N., Ptak, J., Szabo, S., … & Velculescu, V. E. (2004). High frequency of mutations of the PIK3CA gene in human cancers. Science, 304(5670), 554.
https://doi.org/10.1126/science.1096502
Whitman, M., Downes, C. P., Keeler, M., Keller, T., & Cantley, L. (1988). Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature, 332(6165), 644–646.
https://doi.org/10.1038/332644a0
Fruman, D. A., Chiu, H., Hopkins, B. D., Bagrodia, S., Cantley, L. C., & Abraham, R. T. (2017). The PI3K pathway in human disease. Cell, 170(4), 605–635.
https://doi.org/10.1016/j.cell.2017.07.029
Manning, B. D., & Toker, A. (2017). AKT/PKB signaling: Navigating the network. Cell, 169(3), 381–405.
https://doi.org/10.1016/j.cell.2017.04.001
Vanhaesebroeck, B., Stephens, L., & Hawkins, P. (2012). PI3K signalling: The path to discovery and understanding. Nature Reviews Molecular Cell Biology, 13(3), 195–203.
https://doi.org/10.1038/nrm3290
Houslay, D. M., Anderson, K. E., Chessa, T. A. M., Kulkarni, S., Fritsch, R., Downward, J., & Stephens, L. R. (2016). Coincident signals from GPCRs and receptor tyrosine kinases are uniquely transduced by PI3Kβ in myeloid cells. Science Signaling, 9(441), ra82.
https://doi.org/10.1126/scisignal.aaf6705
Chalhoub, N., & Baker, S. J. (2009). PTEN and the PI3-kinase pathway in cancer. Annual Review of Pathology: Mechanisms of Disease, 4, 127–150.
https://doi.org/10.1146/annurev.pathol.4.110807.092311
Kurek, K. C., Luks, V. L., Ayturk, U. M., Alomari, A. I., Fishman, S. J., Spencer, S. A., … & Warman, M. L. (2012). Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. American Journal of Human Genetics, 90(6), 1108–1115.
https://doi.org/10.1016/j.ajhg.2012.05.006
Lucas, C. L., Zhang, Y., Venida, A., MacNamara, K. C., Wakeland, E. K., Fortner, K. A., … & Candotti, F. (2014). Autoimmunity caused by PIK3CD activating mutations in humans with short stature and immune deficiency. The Journal of Experimental Medicine, 211(12), 2537–2547.
https://doi.org/10.1084/jem.20141157
Elkabets, M., Vora, S., Juric, D., Morse, N., Mino-Kenudson, M., Muranen, T., … & Baselga, J. (2013). mTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. Science Translational Medicine, 5(196), 196ra99.
https://doi.org/10.1126/scitranslmed.3005402
Fruman, D. A., & Cantley, L. C. (2014). Idelalisib—A PI3Kδ inhibitor for B-cell cancers. New England Journal of Medicine, 370(11), 1061–1062.
https://doi.org/10.1056/NEJMe1401460
Hopkins, B. D., Goncalves, M. D., & Cantley, L. C. (2016). Obesity and cancer: Mechanistic insights and perspectives. Cell, 166(5), 1148–1160.
https://doi.org/10.1016/j.cell.2016.08.023
Brastianos, P. K., Carter, S. L., Santagata, S., Cahill, D. P., Taylor-Weiner, A., Jones, R. T., … & Johnson, B. E. (2015). Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discovery, 5(11), 1164–1177.
https://doi.org/10.1158/2159-8290.CD-15-0369
Peng, W., Chen, J. Q., Liu, C., Malu, S., Creasy, C., Tetzlaff, M. T., … & Hwu, P. (2016). Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discovery, 6(2), 202–216.