The Evolution of Bcr-Abl Inhibitors: From Imatinib to Next-Gen Therapies for CML and Beyond
Abstract
The advent of Bcr-Abl inhibitors has revolutionized the treatment landscape of chronic myeloid leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL). This blog post explores the historical development and structural evolution of these targeted therapies, beginning with imatinib, the first tyrosine kinase inhibitor (TKI) designed to selectively block the Bcr-Abl fusion protein. As resistance mutations—particularly T315I—emerged, second- and third-generation inhibitors such as nilotinib, dasatinib, and ponatinib were rationally designed to overcome therapeutic failure. The discussion further highlights promising preclinical compounds and emerging strategies, including allosteric inhibitors and combination regimens. Finally, this article reflects on broader translational insights gained from the Bcr-Abl inhibitor model, emphasizing its relevance to precision oncology and future drug development across diverse malignancies.
The Philadelphia Chromosome and the Birth of Targeted Cancer Therapy
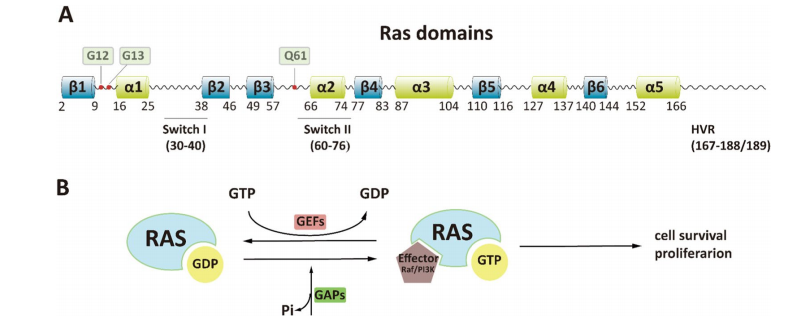
The discovery of the Philadelphia chromosome marked a paradigm shift in our understanding of cancer biology and treatment. This abnormal chromosome, formed by a translocation between chromosomes 9 and 22 (t(9;22)), leads to the creation of the Bcr-Abl fusion gene. This fusion encodes a constitutively active tyrosine kinase that drives uncontrolled proliferation of hematopoietic cells, making it the central oncogenic driver in chronic myeloid leukemia (CML) and a subset of acute lymphoblastic leukemia (ALL) cases.
Unlike traditional chemotherapies that non-selectively target dividing cells, the Bcr-Abl fusion provided a clear molecular target. The development of imatinib mesylate (also known as STI571) was a pioneering moment in oncology, as it was the first drug specifically designed to inhibit a cancer-causing tyrosine kinase. Imatinib functions by binding to the ATP-binding site of Bcr-Abl in its inactive (DFG-out) conformation, effectively halting its signaling cascade and inducing apoptosis in leukemic cells.
The success of imatinib not only transformed the prognosis for CML patients—dramatically increasing survival and quality of life—but also ignited the era of targeted therapy. It demonstrated for the first time that a small molecule could selectively inhibit a cancer-specific protein with manageable side effects. This breakthrough validated the concept of molecular precision in cancer therapy, comparable to the earlier hormonal targeting in breast cancer with tamoxifen.
Furthermore, imatinib became a template for rational drug design, combining high-throughput screening, structural biology, and structure-activity relationship analysis. It laid the groundwork for the development of second- and third-generation inhibitors, which were designed to overcome resistance mutations.
In sum, the identification of the Bcr-Abl oncoprotein and the subsequent development of imatinib exemplify how molecular oncology can translate basic discoveries into life-changing therapies, and how a single chromosomal abnormality can become the Achilles’ heel of a malignancy.
Imatinib and the Structural Blueprint of First-Generation TKIs
Imatinib mesylate (STI571) marked a groundbreaking advancement in cancer pharmacology as the first successful small-molecule inhibitor targeting a specific oncogenic kinase. Developed in the 1990s, imatinib was designed to inhibit the ATP-binding site of the Bcr-Abl tyrosine kinase, a fusion protein produced by the Philadelphia chromosome translocation. This structural abnormality drives the pathogenesis of chronic myeloid leukemia (CML) and some forms of acute lymphoblastic leukemia (ALL).
Imatinib’s structure is engineered to exploit the inactive, or DFG-out, conformation of the Bcr-Abl kinase domain. It binds tightly to the ATP-binding cleft, stabilizing the inactive conformation and preventing phosphorylation of downstream signaling proteins. The drug interacts with key residues within the kinase domain—particularly the “gatekeeper” threonine at position 315 (Thr315)—via a combination of hydrogen bonds and Van der Waals interactions. This mode of binding ensures selectivity for Bcr-Abl over other kinases while maintaining potent inhibitory activity.
The core scaffold of imatinib, a phenylaminopyrimidine, was modified through medicinal chemistry to improve its pharmacokinetics, oral bioavailability, and aqueous solubility. Strategic incorporation of groups such as N-methylpiperazine and a spacer benzene ring reduced mutagenic potential while enhancing target affinity. The final compound displayed remarkable efficacy in vitro and in clinical settings, leading to its approval in 2001.
Despite its success, resistance to imatinib emerged in nearly one-third of patients, prompting in-depth structural investigations. Most resistance was traced to point mutations within the Bcr-Abl kinase domain, particularly the T315I mutation, which disrupts a critical hydrogen bond and prevents the conformational shift required for imatinib binding. These findings underscored the importance of structure-activity relationship (SAR) studies in understanding and overcoming therapeutic resistance.
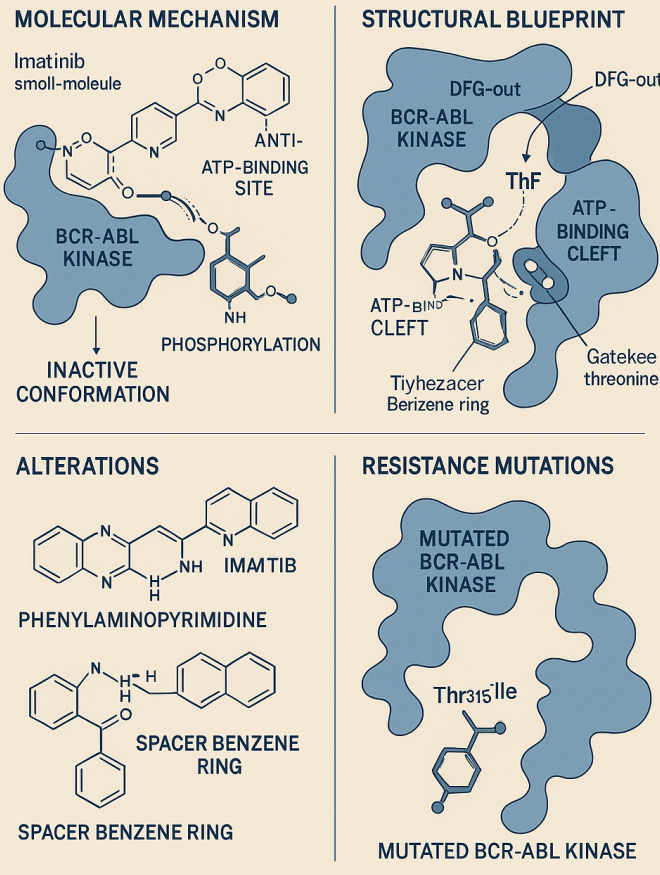
Fig1. Imatinib: Structure, Mechanism, and Resistance
Imatinib’s development has since served as a blueprint for rational drug design, setting the stage for second- and third-generation TKIs that address resistance and broaden the therapeutic scope. Its success exemplifies how structural biology can guide targeted therapy to combat complex malignancies like CML.
Second-Generation TKIs – Meeting the Challenge of Resistance
Although imatinib transformed chronic myeloid leukemia (CML) into a manageable disease, resistance—particularly due to point mutations in the Bcr-Abl kinase domain—necessitated the development of second-generation tyrosine kinase inhibitors (TKIs). These newer agents were engineered to bind more flexibly or potently to mutant forms of Bcr-Abl while preserving selectivity.
Nilotinib (AMN107), structurally related to imatinib, features modifications such as an inverted amide linker and a trifluoromethyl group to increase van der Waals interactions. These changes reduce the drug’s dependence on hydrogen bonding, enabling activity against many imatinib-resistant mutations. Nilotinib exhibits higher potency in vitro (IC₅₀: 10–25 nM vs. 100–500 nM for imatinib), but it is ineffective against the T315I mutation. A notable advantage of nilotinib is its independence from drug transporters, making it less vulnerable to resistance via altered drug efflux or uptake.
Dasatinib (BMS-354825), in contrast, targets both active (DFG-in) and inactive (DFG-out) conformations of Bcr-Abl. With a smaller and more flexible molecular structure, dasatinib binds in multiple conformational states and can overcome several imatinib-resistant mutations—except T315I. Clinical trials have shown that dasatinib produces faster and deeper molecular responses in newly diagnosed CML patients compared to imatinib. This dual-binding flexibility represents a novel approach in kinase inhibitor design.
Bosutinib (SKI-606), originally developed as a Src inhibitor, also inhibits Bcr-Abl but with less specificity. While ineffective against T315I and some P-loop mutations, it offers clinical utility due to its resistance to efflux by multidrug transporters and fewer off-target effects.
Despite improvements, these second-generation TKIs still fall short against the T315I mutation, often dubbed the “gatekeeper” mutation due to its ability to block drug binding through steric hindrance and conformational stabilization. This limitation ultimately paved the way for third-generation inhibitors specifically designed to address this mutation.
In summary, nilotinib, dasatinib, and bosutinib significantly expanded the treatment arsenal for CML, demonstrating how structural refinement can extend drug utility and address emerging resistance mechanisms.
Ponatinib and Emerging Bcr-Abl Inhibitors for Refractory Mutations
Among the most formidable obstacles in chronic myeloid leukemia (CML) treatment is the T315I mutation in the Bcr-Abl kinase domain—often referred to as the “gatekeeper” mutation. This single amino acid substitution (threonine to isoleucine) blocks a critical hydrogen bond and stabilizes the active conformation of the kinase, rendering both first- and second-generation TKIs largely ineffective. To overcome this challenge, ponatinib, the first third-generation TKI, was specifically engineered with this mutation in mind.
Ponatinib’s structure features an ethynyl linkage that allows it to circumvent steric hindrance posed by the bulky isoleucine residue at position 315. Unlike its predecessors, ponatinib can bind to the DFG-out conformation even in the presence of T315I. It maintains high binding affinity through a network of hydrogen bonds and van der Waals interactions, showing low nanomolar IC₅₀ values across a spectrum of Bcr-Abl mutants, including compound mutations. Clinically, ponatinib has demonstrated significant efficacy in patients with T315I-positive CML or Ph+ ALL who have failed prior therapies.
Beyond ponatinib, several emerging inhibitors have shown promise in preclinical or early clinical trials. Bafetinib and rebastinib aim to refine selectivity and minimize off-target effects. Tozasertib and danusertib, which also target Aurora kinases, display activity against T315I by binding to the active conformation of Bcr-Abl. Another exciting class includes GNF-2 and GNF-5, which bind to the allosteric myristoyl site, inducing conformational changes that render Bcr-Abl inactive. When combined with ATP-competitive inhibitors like dasatinib or nilotinib, they exhibit synergistic effects, overcoming otherwise resistant clones.
While many of these newer compounds have yet to achieve regulatory approval, their development represents a strategic diversification of therapeutic mechanisms—targeting not only the ATP-binding site but also alternative conformations and regulatory domains. Together, these innovations mark a move toward precision medicine for patients with resistant forms of leukemia.
The Future of Bcr-Abl Therapy and Broader Lessons in Oncology
The development of Bcr-Abl inhibitors, particularly imatinib and its successors, represents a textbook case of translational medicine—transforming molecular insights into life-saving therapies. Yet, as the clinical landscape evolves, so too must the strategies to combat resistance and disease heterogeneity in chronic myeloid leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL).
One of the next frontiers is addressing compound mutations—patients harboring multiple Bcr-Abl mutations within the same leukemic clone. These complex profiles pose a significant challenge, as even potent inhibitors like ponatinib may fail to fully suppress resistant subpopulations. The need for combination therapies, such as pairing ATP-competitive inhibitors with allosteric modulators like GNF-2 or GNF-5, is becoming increasingly apparent. Such approaches can exploit multiple binding sites or conformational states of Bcr-Abl, improving efficacy even in the face of mutational diversity.
Beyond kinase inhibition, future therapies are likely to adopt multifaceted strategies. These include targeting parallel oncogenic pathways or microenvironmental factors that support leukemic survival. Experimental agents such as ALT-803 (an IL-15 superagonist), panobinostat (an HDAC inhibitor), and rigosertib (a RAS pathway antagonist) exemplify a shift toward modulating broader signaling networks.
Another promising direction lies in reverse translational research—where clinical observations of resistance or relapse guide new hypotheses for bench-side exploration. This iterative loop between lab and clinic has already led to a more refined understanding of differential signaling between Bcr-Abl isoforms (e.g., p190 vs. p210), which could influence future drug development.
Importantly, the lessons from Bcr-Abl inhibition extend far beyond leukemia. Similar resistance mechanisms have been observed in EGFR-mutant lung cancers and ALK-rearranged lymphomas, making the strategic framework established by Bcr-Abl therapies broadly applicable.
Ultimately, the trajectory of Bcr-Abl inhibitor development underscores the power of precision oncology, not just in designing smart drugs but in adapting them dynamically to outpace tumor evolution. As technologies like proteomics, genomics, and structural modeling advance, the horizon for targeted cancer therapy grows ever broader.
References
Druker, B. J., Tamura, S., Buchdunger, E., et al. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nature Medicine, 2(5), 561–566.
https://doi.org/10.1038/nm0596-561
Rossari, F., Minutolo, F., & Orciuolo, E. (2018). Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. Journal of Hematology & Oncology, 11(1), 84.
https://doi.org/10.1186/s13045-018-0624-2
Ward, H. W. (1973). Anti-oestrogen therapy for breast cancer: a trial of tamoxifen at two dose levels. British Medical Journal, 1(5844), 13–14.
https://doi.org/10.1136/bmj.1.5844.13
O’Brien, S. G., Guilhot, F., Larson, R. A., et al. (2003). Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. New England Journal of Medicine, 348(11), 994–1004.
https://doi.org/10.1056/NEJMoa022457
Schindler, T., Bornmann, W., Pellicena, P., et al. (2000). Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science, 289(5486), 1938–1942.
https://doi.org/10.1126/science.289.5486.1938
Capdeville, R., Buchdunger, E., Zimmermann, J., & Matter, A. (2002). Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nature Reviews Drug Discovery, 1(7), 493–502.
https://doi.org/10.1038/nrd839
Azam, M., Seeliger, M. A., Gray, N. S., Kuriyan, J., & Daley, G. Q. (2008). Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nature Structural & Molecular Biology, 15(10), 1109–1118.
https://doi.org/10.1038/nsmb.1486
Weisberg, E., Manley, P. W., Breitenstein, W., et al. (2005). Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell, 7(2), 129–141.
https://doi.org/10.1016/j.ccr.2005.01.007
Shah, N. P., Tran, C., Lee, F. Y., et al. (2004). Overriding imatinib resistance with a novel ABL kinase inhibitor. Science, 305(5682), 399–401.
https://doi.org/10.1126/science.1099480
Cortes, J. E., Gambacorti-Passerini, C., Deininger, M. W., et al. (2017). Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: Results from the randomized BFORE trial. Journal of Clinical Oncology, 36(3), 231–237.
https://doi.org/10.1200/JCO.2017.74.7162
Rossari, F., Minutolo, F., & Orciuolo, E. (2018). Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. Journal of Hematology & Oncology, 11(1), 84.
https://doi.org/10.1186/s13045-018-0624-2
Cortes, J. E., Kantarjian, H., Shah, N. P., et al. (2012). Ponatinib in refractory Philadelphia chromosome–positive leukemias. New England Journal of Medicine, 367(22), 2075–2086.
https://doi.org/10.1056/NEJMoa1205127
Deininger, M. W., Hodgson, J. G., Shah, N. P., et al. (2016). Compound mutations in BCR-ABL1 are not major drivers of primary or secondary resistance to ponatinib in CP-CML patients. Blood, 127(6), 703–712.
https://doi.org/10.1182/blood-2015-09-671719