Targeting RAS Mutations in Cancer: From Undruggable to Unstoppable Innovation
Abstract
RAS mutations represent one of the most frequent and challenging oncogenic drivers in human cancers, historically considered “undruggable” due to their structural and biochemical properties. However, recent advances have revolutionized this landscape. This blog explores the biology of RAS signaling, its clinical implications, and the emergence of targeted therapies such as KRAS G12C inhibitors, SHP2 blockers, and RNA-based approaches. By highlighting both breakthroughs and ongoing challenges—such as drug resistance and mutation-specific treatment limitations—this article emphasizes the need for personalized, combination-based strategies to treat RAS-driven malignancies. As precision oncology continues to evolve, RAS-targeted therapy stands at the forefront of cancer innovation.
Targeting the Undruggable: Why RAS Mutations Remain a Major Cancer Challenge
Mutations in the RAS gene family represent one of the most pressing and persistent challenges in cancer research and treatment. Found in approximately 19% of all human cancers, RAS mutations are strongly associated with aggressive tumor behavior, treatment resistance, and poor prognosis (Chen et al., 2021). Despite being discovered over four decades ago, RAS has long remained elusive to drug developers, earning a reputation as “undruggable.”
The RAS family consists of three proto-oncogenes: KRAS, NRAS, and HRAS, which encode small GTPase proteins that regulate key signaling pathways controlling cell growth, differentiation, and survival. Under normal physiological conditions, RAS proteins act like switches, cycling between an active GTP-bound state and an inactive GDP-bound state. However, when mutated, they become stuck in a permanently “on” state, fueling uncontrolled proliferation and survival of cancer cells (Cox et al., 2014).
Among the RAS isoforms, KRAS is the most frequently mutated, particularly in lung adenocarcinoma, colorectal cancer, and pancreatic ductal adenocarcinoma. Specific hotspot mutations—such as G12C, G12D, and Q61K—disrupt the GTPase activity of RAS, locking it into its active form. These mutations not only drive tumor initiation and maintenance but also contribute to resistance against standard therapies, including chemotherapy and targeted agents like EGFR inhibitors (Prior et al., 2020).
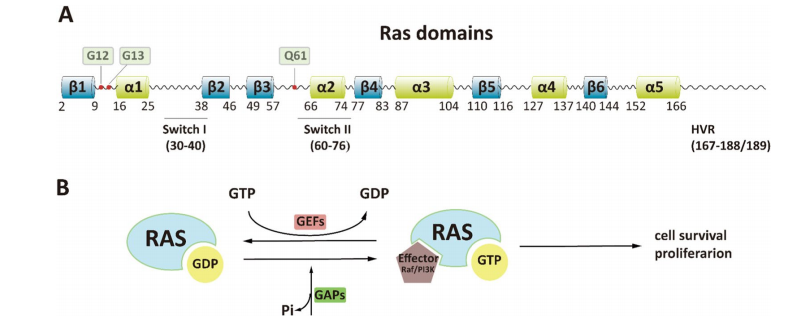
Fig. 1 Structure and switch of RAS.
Until recently, efforts to directly inhibit RAS proteins had failed due to their smooth surface and high affinity for GTP/GDP, which left little room for small molecules to bind effectively. However, recent breakthroughs—most notably the FDA approval of sotorasib, a covalent inhibitor targeting KRAS G12C—have reignited hope in the fight against RAS-driven cancers (Canon et al., 2019).
As we stand on the cusp of a new era in oncology, understanding why RAS matters—and how we can finally drug it—has become more urgent than ever. In this series, we’ll explore the biology, clinical implications, and the most promising strategies for tackling RAS mutations in cancer.
Decoding RAS: How This Small Protein Fuels Big Problems in Cancer
To understand why RAS mutations are so formidable in cancer, we first need to explore the structure and function of RAS proteins—and how subtle changes at the molecular level can drive devastating biological consequences.
The RAS family of genes includes KRAS, NRAS, and HRAS, which produce four major protein isoforms: KRAS4A, KRAS4B, NRAS, and HRAS. All RAS proteins function as GTPases, molecular switches that toggle between an “on” (GTP-bound) and “off” (GDP-bound) state. This cycle regulates several essential cell processes: proliferation, differentiation, migration, and survival (Vetter & Wittinghofer, 2001).
Structurally, RAS proteins share a conserved G-domain (amino acids 1–166) and a hypervariable C-terminal region that helps anchor them to the cell membrane. The G-domain includes regions known as Switch I and Switch II, which are critical for binding to downstream effectors and activating signaling cascades such as the MAPK (RAF–MEK–ERK) and PI3K–AKT–mTOR pathways (Cox et al., 2014).
Under normal conditions, RAS is activated in response to extracellular signals via guanine nucleotide exchange factors (GEFs), which promote GTP binding. GTPase-activating proteins (GAPs) then turn off the signal by accelerating GTP hydrolysis. However, oncogenic mutations in key codons—G12, G13, and Q61—cripple RAS’s ability to hydrolyze GTP, locking it into a constitutively active state (Simanshu & Cox, 2017). This leads to persistent downstream signaling, promoting uncontrolled cell growth and resistance to apoptosis.
Importantly, the type and location of the mutation often dictate the cancer’s behavior. For example, KRAS G12C is common in lung adenocarcinoma, while NRAS Q61 mutations are frequently found in melanoma. These differences also influence how tumors respond—or don’t respond—to targeted therapies.
RAS’s small size and lack of deep binding pockets make it a challenging target for drug design. But as research uncovers more about its structural nuances, especially mutation-specific conformations, new therapeutic opportunities are emerging.
Ultimately, RAS is a master regulator gone rogue in many cancers. Understanding its structure and function is key to designing therapies that can finally shut it down.
RAS Mutations in the Clinic: Predicting Resistance and Guiding Cancer Therapy
While much is known about the molecular role of RAS mutations, their clinical implications are equally significant. Mutations in KRAS, NRAS, or HRAS not only drive tumor development but also influence tumor aggressiveness, treatment response, and patient prognosis across several cancer types.
For example, in colorectal cancer (CRC), KRAS mutations—particularly in codons 12 and 13—are linked to poor differentiation, mucinous histology, and lung metastasis rather than liver metastasis, which is more common in KRAS wild-type tumors (Tie et al., 2011). Importantly, KRAS mutations predict resistance to anti-EGFR monoclonal antibodies such as cetuximab and panitumumab. Patients with these mutations do not benefit from these therapies, and using them in such cases can lead to unnecessary toxicity and cost (Douillard et al., 2013).
In non-small cell lung cancer (NSCLC), the role of KRAS mutations is more nuanced. Some studies suggest that KRAS-mutant tumors are less responsive to EGFR-targeted tyrosine kinase inhibitors (TKIs) like erlotinib. However, KRAS status has not been consistently validated as a predictive biomarker for TKI therapy in NSCLC, making it a subject of ongoing debate (Zer et al., 2016).
RAS mutations also influence the efficacy of immunotherapy. In KRAS-mutant NSCLC, tumors often express higher levels of PD-L1, a key checkpoint molecule, suggesting better responses to immune checkpoint blockade (ICB). Indeed, studies have shown that patients with KRAS mutations may experience greater benefit from PD-1/PD-L1 inhibitors (Jeanson et al., 2019). However, in other cancers like colorectal cancer, RAS mutations have been associated with immune evasion and poor immunotherapy responses, especially when combined with other mutations like STK11 or PTEN loss (Liao et al., 2019).
Ultimately, RAS mutation status has become an essential biomarker in personalized cancer treatment. It helps oncologists select or avoid specific therapies, anticipate resistance, and tailor combination strategies. As clinical research evolves, the role of RAS will continue to expand—not just as a molecular villain, but as a therapeutic guide.
Breaking the Barrier: Innovative Therapies Targeting RAS in Cancer
For decades, RAS proteins were labeled “undruggable” due to their smooth molecular surfaces and picomolar affinity for GTP, which made it extremely difficult to design inhibitors that could bind effectively. However, recent breakthroughs in drug discovery and structural biology have shattered that perception, leading to the development of novel direct and indirect strategies to target RAS-driven cancers.
The most groundbreaking advance to date is the development of KRAS G12C-specific inhibitors, such as sotorasib (AMG510) and adagrasib (MRTX849). These small molecules exploit a newly discovered binding pocket created by the G12C mutation and irreversibly lock KRAS in its inactive GDP-bound form, effectively shutting down its oncogenic signaling. In 2021, sotorasib became the first FDA-approved drug targeting a RAS mutation, marking a historic milestone in cancer therapy. Clinical trials have shown significant activity in non-small cell lung cancer (NSCLC), with disease control rates exceeding 80% (Canon et al., 2019).
Beyond G12C, researchers are developing next-generation inhibitors for other KRAS mutations, such as G12D and G13D, which are prevalent in colorectal and pancreatic cancers. For example, MRTX1133, a non-covalent KRAS G12D inhibitor, has demonstrated promising preclinical activity. These agents, still in early development, represent hope for expanding the range of patients who can benefit from mutation-specific RAS inhibition.
In parallel, indirect targeting strategies are gaining traction. These include:
SHP2 inhibitors (e.g., RMC-4630), which block upstream signaling that activates RAS;
SOS1 inhibitors that prevent RAS activation by inhibiting guanine nucleotide exchange;
Combination therapies, such as pairing MEK inhibitors with PI3K or SHP2 inhibitors, to overcome resistance mechanisms and enhance treatment efficacy.
Another promising approach involves RNA-based therapies, such as antisense oligonucleotides and siRNA, which suppress RAS expression at the mRNA level. Innovative delivery systems—like nanoparticles and exosomes—are being developed to overcome barriers in stability and tumor targeting.
While no single approach offers a universal solution, the field is rapidly advancing. A multi-pronged strategy that targets RAS directly, disrupts its regulatory network, and leverages synthetic lethality is emerging as the new gold standard in RAS-targeted cancer therapy.
Looking Ahead: Personalized Strategies for RAS-Driven Cancers
As our understanding of RAS signaling and tumor biology deepens, the future of cancer therapy is shifting toward precision strategies tailored to individual mutations and tumor contexts. While the approval of KRAS G12C inhibitors has transformed the outlook for a subset of patients, much work remains to extend these advances to the broader landscape of RAS-driven cancers.
One of the greatest challenges ahead is overcoming drug resistance. Even in patients who initially respond to KRAS G12C inhibitors like sotorasib or adagrasib, resistance frequently develops within months. Mechanisms include secondary KRAS mutations (e.g., Y96D or H95R), bypass pathway activation (e.g., upregulation of NRAS, BRAF, or MET), and phenotypic changes, such as epithelial-to-mesenchymal transition or histological transformation (Awad et al., 2021). To combat this, future regimens will likely rely on combination therapies—such as pairing KRAS inhibitors with SHP2, MEK, or EGFR inhibitors—to delay or prevent resistance.
Personalized therapy also means expanding the arsenal beyond G12C. Researchers are rapidly developing inhibitors for KRAS G12D, G13D, and Q61 mutations, particularly relevant in pancreatic and colorectal cancers. In parallel, biomarker-driven stratification—based on co-occurring mutations (e.g., STK11, TP53, or KEAP1)—can help predict therapeutic response and guide treatment decisions.
Another promising avenue is immunotherapy. Although KRAS mutations are traditionally associated with an immunosuppressive tumor microenvironment, emerging data suggest that specific KRAS variants may correlate with high PD-L1 expression or tumor mutational burden, especially in lung cancer. Clinical trials are now exploring personalized KRAS neoantigen vaccines and adoptive T cell therapies targeting mutation-specific epitopes.
Cutting-edge platforms such as CRISPR-based synthetic lethality screens and machine learning models are also helping identify vulnerabilities unique to RAS-mutant tumors. These efforts are driving the discovery of context-specific dependencies that can be exploited with next-generation targeted agents.
Ultimately, the fight against RAS-driven cancers will not be won by a single “magic bullet,” but by integrating genomics, immunology, and systems biology into dynamic, personalized treatment plans. With collaboration between academia, biotech, and clinical oncology, the once “undruggable” RAS is becoming a gateway to a new era of precision cancer medicine.
References
Canon, J., Rex, K., Saiki, A. Y., Mohr, C., Cooke, K., Bagal, D., … & Olson, P. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature, 575(7781), 217–223.
https://doi.org/10.1038/s41586-019-1694-1
Chen, Y., Ye, Z., Huang, Y., Zhou, D., & Wang, Y. (2021). Emerging strategies to target RAS signaling in human cancer therapy. Journal of Hematology & Oncology, 14(1), 1–19.
https://doi.org/10.1186/s13045-021-01127-w
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J., & Der, C. J. (2014). Drugging the undruggable RAS: Mission possible? Nature Reviews Drug Discovery, 13(11), 828–851.
https://doi.org/10.1038/nrd4389
Prior, I. A., Hood, F. E., & Hartley, J. L. (2020). The frequency of Ras mutations in cancer. Cancer Research, 80(14), 2969–2974.
https://doi.org/10.1158/0008-5472.CAN-19-3682
Simanshu, D. K., & Cox, A. D. (2017). Structural insights into RAS regulation and function. Current Opinion in Cell Biology, 45, 102–109.
https://doi.org/10.1016/j.ceb.2017.03.002
Vetter, I. R., & Wittinghofer, A. (2001). The guanine nucleotide-binding switch in three dimensions. Science, 294(5545), 1299–1304.
https://doi.org/10.1126/science.1062023
Douillard, J. Y., Oliner, K. S., Siena, S., Tabernero, J., Burkes, R., Barugel, M., … & Sidhu, R. (2013). Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. New England Journal of Medicine, 369(11), 1023–1034.
https://doi.org/10.1056/NEJMoa1305275
Jeanson, A., Tomasini, P., Souquet-Bressand, M., Brandone, N., Boucekine, M., Grangeon, M., … & Barlesi, F. (2019). Efficacy of immune checkpoint inhibitors in KRAS-mutant non-small cell lung cancer (NSCLC). Journal of Thoracic Oncology, 14(6), 1095–1101.
https://doi.org/10.1016/j.jtho.2019.02.009
Liao, W., Overman, M. J., Boutin, A. T., Shang, X., Zhao, D., Dey, P., … & Wang, H. (2019). KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell, 35(4), 559–572.e7.
https://doi.org/10.1016/j.ccell.2019.02.008
Tie, J., Lipton, L., Desai, J., Gibbs, P., Jorissen, R. N., Christie, M., … & Phillips, W. A. (2011). KRAS mutation is associated with lung metastasis in patients with curatively resected colorectal cancer. Clinical Cancer Research, 17(5), 1122–1130.