Targeting Focal Adhesion Kinase in Cancer: Advances in Inhibitors and PROTAC-Based Degraders
Abstract
Focal Adhesion Kinase (FAK) is a non-receptor tyrosine kinase with dual roles as a signaling enzyme and scaffolding protein, contributing to cancer development, metastasis, and therapy resistance. Although several FAK inhibitors have entered clinical trials, they primarily target kinase activity and fail to eliminate scaffolding functions. Proteolysis-targeting chimeras (PROTACs) offer a novel approach by degrading the entire FAK protein, thereby simultaneously disabling both functional domains. Recent advances have led to the development of potent and selective FAK-targeting PROTACs, demonstrating promising anti-proliferative and anti-metastatic effects in preclinical models. This review summarizes FAK structure, clinical progress of small-molecule inhibitors, and the emerging potential of PROTAC-based FAK degraders as next-generation cancer therapeutics.
Introduction
Focal Adhesion Kinase (FAK) is a non-receptor tyrosine kinase that functions both as an enzyme and a scaffold. Structurally, it comprises a FERM domain, a kinase domain, a proline-rich region, and a FAT domain. FAK interacts with over 50 proteins and plays a pivotal role in cancer development, metastasis, and recurrence. Although eight FAK inhibitors have advanced to clinical trials, they primarily target its kinase activity, leaving the scaffolding function intact. Proteolysis-targeting chimera (PROTAC) technology offers a novel approach by promoting the degradation of the entire FAK protein, thereby simultaneously eliminating both its enzymatic and scaffolding functions. Several FAK-targeting PROTACs have been developed, showing promising therapeutic potential. This article reviews the current landscape of FAK inhibitors and protein degraders.
FAK Kinase Background
Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase first discovered by Guan and colleagues in 1991. FAK is encoded by a gene located on human chromosome 8 and is able to interact with receptors on the plasma membrane as well as various protein complexes in the nucleus.
The human Focal Adhesion Kinase (FAK) protein is composed of several distinct structural domains: an N-terminal FERM domain, a central kinase domain, three proline-rich regions (PRI, PRII, PRIII), and a C-terminal FAT domain. The FERM domain mediates interactions with the intracellular regions of transmembrane receptors, anchoring FAK to membrane-associated signaling complexes. The kinase domain, spanning amino acid residues 390 to 650, is highly conserved and contains at least six tyrosine residues that are essential for downstream FAK signaling. At the C-terminus, the FAT domain facilitates binding to key focal adhesion proteins, including paxillin, talin, and vascular endothelial growth factor receptor 3 (VEGFR3), playing a crucial role in cell adhesion and signaling dynamics.
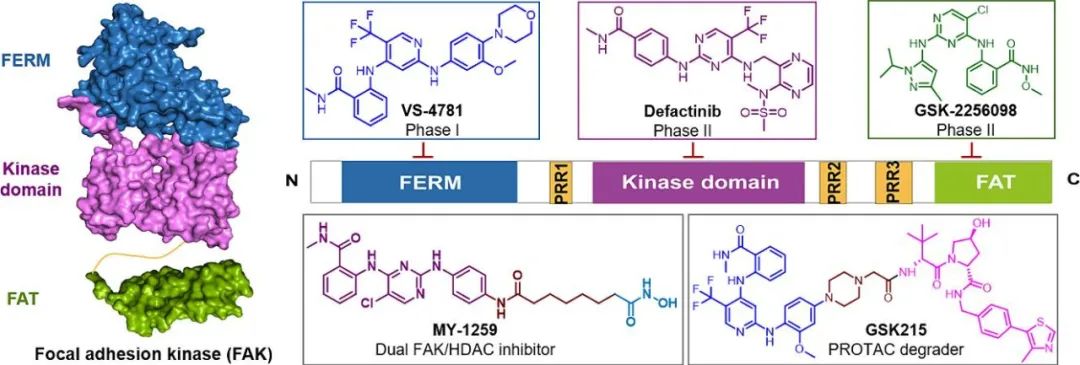
Figure 1. Schematic diagram of the structure of FAK.
Following integrin activation, Focal Adhesion Kinase (FAK) undergoes autophosphorylation in a kinase-dependent manner, primarily at the Tyr397 residue. This phosphorylation event promotes FAK dimerization and triggers the activation of multiple downstream signaling pathways that regulate critical cellular processes such as proliferation, migration, and invasion. Beyond its enzymatic function, FAK also serves as a scaffolding hub at the plasma membrane, assembling large multi-protein signaling complexes. With over 50 known binding sites, FAK orchestrates both catalytic and structural roles in signal transduction. Its central involvement in cancer progression, metastasis, and recurrence has led to its emergence as a promising therapeutic target in oncology.
FAK inhibitors
Currently, eight FAK inhibitors have entered clinical trials (Figure 2). Notably, combination therapies involving FAK inhibitors have shown good clinical effects.
Ifebemtinib, developed by InxMed, is an ATP-competitive FAK inhibitor that is primarily used to treat solid tumors, including platinum-resistant ovarian cancer. At the 2024 American Society of Clinical Oncology (ASCO 2024) Annual Meeting, the results of a Phase Ib/II clinical study of Ifebemtinib in combination with the KRAS G12C inhibitor D-1553 (garsorasib) were announced. Among 31 evaluable patients, the objective response rate (ORR) reached 90.3% and the disease control rate (DCR) was 96.8%, highlighting the potential of this dual oral, chemotherapy-free combination regimen in improving the treatment outcomes of patients with KRAS G12C-mutated non-small cell lung cancer (NSCLC). Ifebemtinib has been granted Fast Track designation by the U.S. FDA in August 2021 and Breakthrough Therapy designation by the China National Medical Products Administration (NMPA) in April 2022. A placebo-controlled, randomized, double-blind pivotal trial for platinum-resistant ovarian cancer (PROC) is currently underway. The company plans to submit a new drug application to the NMPA in 2025.
On December 30, 2024, Verastem Oncology announced that the U.S. FDA has accepted a new drug application (NDA) for the combination therapy of avutometinib and defactinib. This acceptance is an accelerated approval procedure for the treatment of adult patients with recurrent low-grade serous ovarian cancer (LGSOC) with KRAS mutations. Once approved, the combination therapy of avutometinib and defactinib will become the first FDA-approved treatment specifically for adult patients with recurrent KRAS mutant LGSOC.

Figure 2. Small molecule inhibitors of FAK in clinical investigation.
Although some FAK inhibitors have shown efficacy in preclinical studies, clinical success has not been observed. In addition, defactinib failed in its initial clinical trial against malignant pleural mesothelioma stem cells. Recent progress has been largely limited to the development of FAK kinase inhibitors, which are unable to block the scaffolding function of FAK. Therefore, essential functions mediated by the scaffolding activity of FAK remain beyond the scope of any kinase inhibitor.
FAK protein degrader
The proteolysis targeted chimera (PROTAC) strategy provides a novel therapeutic approach. This method uses the cell’s own degradation mechanism to selectively degrade the target protein, thereby simultaneously destroying the catalytic and non-enzymatic functions of FAK.
In 2018, Craig M. Crews’ research group reported a PROTACs study targeting FAK, in which PROTAC-3 showed efficient FAK degradation and anti-invasive ability in TNBC cells, and its selectivity was better than defactinib. In 2019, Peter Ettmayer’s team designed and synthesized FAK PROTACs based on the ATP-competitive FAK inhibitor BI-4464. Among them, BI-3663 and BI-0319 showed effective FAK degradation activity, but their anti-proliferative effects were not better than BI-4464. In 2020, the Yu Rao research group reported a FAK PROTACs compound library, in which compound FC-11 showed potent FAK degradation activity. In 2021, the Robert P. Law research group synthesized 6 FAK PROTACs, among which GSK215 showed the strongest FAK degradation activity and effectively inhibited the migration of A549 cells. In 2022, the Maosheng Cheng research group developed a FAK PROTAC based on TAE226, in which compound B5 showed potent FAK degradation, anti-proliferative activity and migration inhibition in A549 lung cancer cells. In 2023, Behnam Nabet’s research group used structure-guided design to develop a highly selective FAK degrader BSJ-04-146, which can significantly degrade FAK, reduce cell viability, and inhibit cell migration at low doses and has good pharmacokinetic properties (Figure 3).
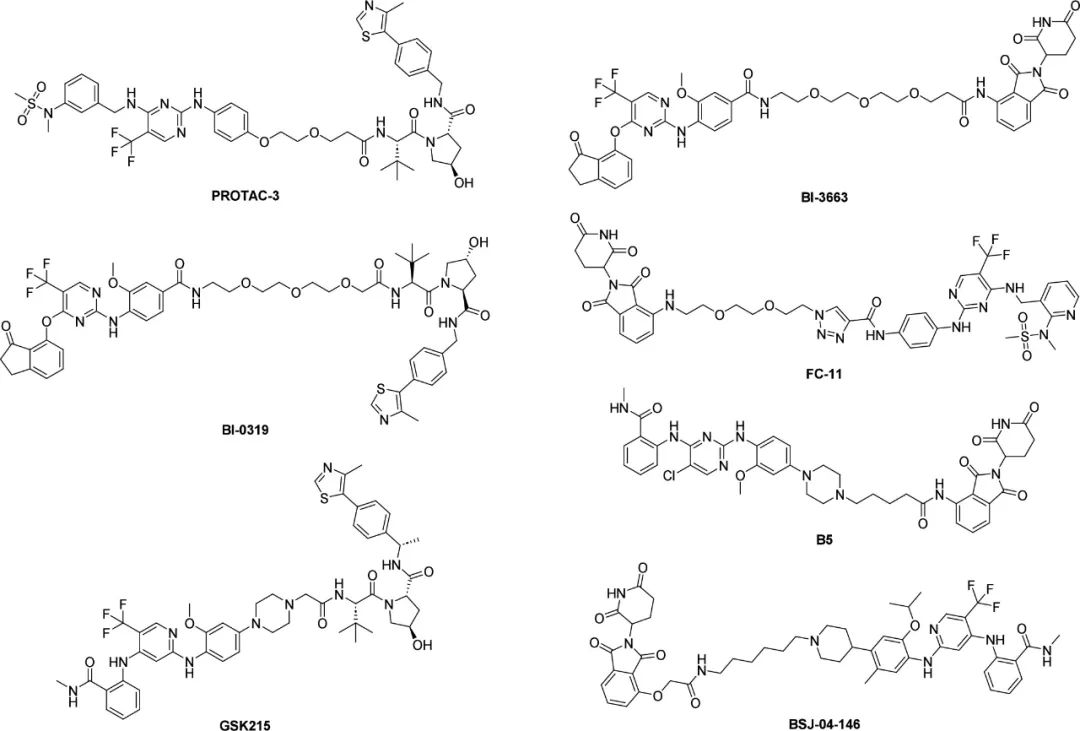
Figure 3. PROTACs targeting FAK.
Future Outlook
Given FAK’s pivotal role in driving tumor progression toward malignant and invasive phenotypes, it has emerged as a highly promising target for anticancer drug development. Combining FAK-targeted therapy with other treatment modalities holds significant potential to enhance therapeutic efficacy, overcome resistance to chemotherapy or targeted agents, and boost the effectiveness of immunotherapy in solid tumors. While combination therapies have shown promise, the development of bifunctional molecules—capable of simultaneously modulating multiple targets—may offer even greater therapeutic benefit. Strategies such as designing dual-target degraders or integrating inhibitory and degradative functions within a single small molecule represent innovative and promising avenues for future cancer treatment. The advancement of FAK degraders into clinical trials is eagerly anticipated, marking a critical step toward translating these discoveries into effective therapies.
References
Cromm PM, Samarasinghe KTG, Hines J, Crews CM. Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J Am Chem Soc. 2018 Dec 12;140(49):17019-17026. doi: 10.1021/jacs.8b08008. Epub 2018 Nov 28. PMID: 30444612.
https://pubs.acs.org/doi/abs/10.1021/jacs.8b08008
Popow J, Arnhof H, Bader G, Berger H, Ciulli A, Covini D, Dank C, Gmaschitz T, Greb P, Karolyi-Özguer J, Koegl M, McConnell DB, Pearson M, Rieger M, Rinnenthal J, Roessler V, Schrenk A, Spina M, Steurer S, Trainor N, Traxler E, Wieshofer C, Zoephel A, Ettmayer P. Highly Selective PTK2 Proteolysis Targeting Chimeras to Probe Focal Adhesion Kinase Scaffolding Functions. J Med Chem. 2019 Mar 14;62(5):2508-2520. doi: 10.1021/acs.jmedchem.8b01826. Epub 2019 Feb 22. PMID: 30739444.
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.8b01826
Law RP, Nunes J, Chung CW, Bantscheff M, Buda K, Dai H, Evans JP, Flinders A, Klimaszewska D, Lewis AJ, Muelbaier M, Scott-Stevens P, Stacey P, Tame CJ, Watt GF, Zinn N, Queisser MA, Harling JD, Benowitz AB. Discovery and Characterisation of Highly Cooperative FAK-Degrading PROTACs. Angew Chem Int Ed Engl. 2021 Oct 18;60(43):23327-23334. doi: 10.1002/anie.202109237. Epub 2021 Sep 17. PMID: 34416073.
https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.202109237
Hu HH, Wang SQ, Shang HL, Lv HF, Chen BB, Gao SG, Chen XB. Roles and inhibitors of FAK in cancer: current advances and future directions. Front Pharmacol. 2024 Feb 12;15:1274209. doi: 10.3389/fphar.2024.1274209. PMID: 38410129; PMCID: PMC10895298.
https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1274209/full