Opportunities and Challenges for Deuteration
Abstract
In the process of drug development, academic employ various methods to enhance the activity and safety of small-molecule compounds, aiming to improve the compound’s other characteristics while maintaining its original activity. Deuterium modification refers to the strategy of replacing specific hydrogen atoms with deuterium atoms within a molecule. As early as the early 1960s, researchers began studying deuterium-modified compounds like d2-tyramine and d3-morphine. Studies have shown that compared to their non-deuterium counterparts, d2-tyramine exhibited superior blood pressure-raising potency, while the potency and toxicity of d3-morphine were reduced. In recent years, deuterium modification has expanded beyond its use in improving the metabolic properties of existing drugs and has become an effective strategy in new drug development.
Deuterium and rationale for drug deuteration
Hydrogen is the most abundant element in the natural world and has three isotopes: protium (1H, abbreviated as H, with 1 proton and no neutrons in the atomic nucleus, abundance of 99.9844%); deuterium (2H, abbreviated as D, with 1 proton and 1 neutron in the atomic nucleus, abundance of 0.0156%); tritium (3H, abbreviated as T, with 1 proton and 2 neutrons in the atomic nucleus, trace amounts). D and T can be detected using mass spectrometry and radioactive measurements, respectively. With the advent of ultra-sensitive mass spectrometry techniques, highly stable deuterium (D) has played a significant role in many biomedical fields. For example, by labeling compounds with deuterium (D), one can track their distribution within the body. By selectively deuterating specific functional groups in drug molecules, the binding process between the drug and its target can be traced, elucidating the biological and chemical processes of the drug in the body.
Compared to hydrogen (H), deuterium (D) has a smaller atomic radius, lower hydrophobicity, shorter C-D bonds compared to C-H bonds, lower vibrational stretching frequency, and lower ground state energy. Deuterium (D) also requires higher free energy for bond dissociation (with a difference of 1.2~1.5 kcal mol⁻¹). As a result, C-D bonds are more stable than C-H bonds. This difference can be quantified using the deuterium kinetic isotope effect (DKIE), where DKIE = kH/kD. A higher DKIE value indicates a slower rate of C-D bond cleavage and greater bond stability.
When the ratio of the rate of C-H bond cleavage to the rate of C-D bond cleavage falls between 1 and 5, it results in a primary deuterium kinetic isotope effect. However, there are some special cases where DKIE is <1 (inverse deuterium kinetic isotope effect) or >5 (large isotope effect, with a theoretical upper limit of 9). Additionally, secondary deuterium kinetic isotope effects arising from neighboring atoms can also affect the cleavage rate, albeit to a lesser extent. When these effects accumulate, they can further slow down the rate of chemical bond cleavage at distant positions.
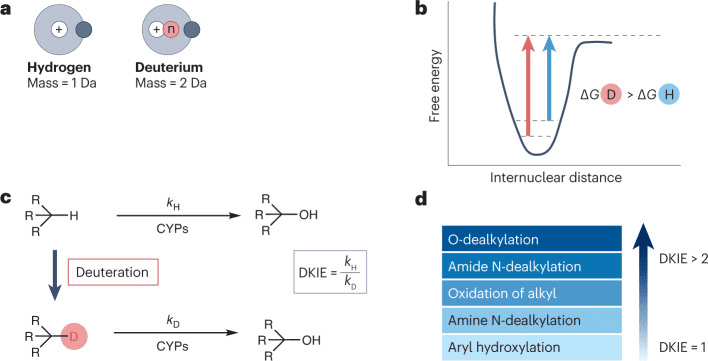
Fig 1. The deuterium kinetic isotope effect and its consequences.[1]
Potential effects of precision deuteration and exemplifying compounds
Conducting deuterium modification at metabolic sites may potentially slow down the metabolism of drugs by CYP450 or other metabolic enzymes (such as alcohol dehydrogenase, aldehyde dehydrogenase, monoamine oxidase, etc.). The magnitude of DKIE depends on the metabolic pathway, the enzymes involved, and the specific substrate. In general, deuterium substitution is most sensitive to O-dealkylation processes, has a significant impact on acylamide N-dealkylation and the oxidation of alkyl groups, has little effect on amine N-dealkylation processes, and does not affect aromatic hydroxylation. Effective deuterium modification can fundamentally alter the drug’s metabolism in the body. For example, it can prolong the drug’s half-life (t1/2), increase the drug’s systemic exposure (area under the curve, AUC), and enhance the maximum blood concentration (maximum concentration), thereby reducing the dosage and dosing frequency required.
However, deuterium modification must be carried out carefully and prudently, as deuterated drugs may undergo metabolism through other unpredicted pathways. This metabolic transformation, known as metabolic switching or metabolic shunting, can potentially result in the overall metabolic characteristics of the final drug remaining unchanged. Therefore, changes in metabolic pathways often require validation through in vivo metabolism experiments.
For instance, the deuterium substitution of a single methyl group of caffeine may promote metabolism at non-deuterated soft sites within the molecule. However, completely deuterating all three methyl groups of caffeine, corresponding to d9-caffeine, significantly inhibits the N-dealkylation process. Interestingly, in a rat model, the peak plasma concentration time of d9-caffeine is delayed compared to caffeine, suggesting that under the cumulative influence of the number of introduced deuterium atoms, the small difference in lipophilicity between D and H can also accumulate, resulting in a slower absorption rate of deuterated drugs by the body. Additionally, a study from 1987 reported that d9-caffeine exhibits significantly increased binding to human serum albumin (HSA) compared to caffeine. This indicates that when developing deuterated drugs, one should also consider the potential issues that this change in binding may pose.
Table 1. Selected examples of deuterated compounds [1]


Possible benefits of deuterium incorporation in drugs
In some application examples, deuterated drugs exhibit distinct advantages in terms of metabolic properties, efficacy, and safety. For instance, the androgen receptor antagonist enzalutamide is primarily hydrolyzed in the body to inactive metabolite M1 and N-demethylated metabolite M2, where M2 may trigger seizures in patients. Deuteration of the N-methyl group to create deutenzalutamide (d3-enzalutamide) improves the metabolic properties, and theoretically reduces side effects. Further head-to-head clinical trials are needed to validate these effects.
In addition, deutivacaftor (d9-ivacaftor), the deuterium product of ivacaftor, and deuterium analogue of the JAK (Janus kinase) inhibitor ruxolitinib (deuruxolitinib, d8-ruxolitinib) also exhibited better metabolic properties with longer t1/2 and higher plasma exposure in vivo. Dextromethorphan hydrobromide is easily metabolized by CYP2D6 enzymes and usually needs to be combined with the potentiator quinidine sulfate (CYP2D6 inhibitor). Deuterated dextromethorphan hydrobromide is significantly less sensitive to CYP2D6 enzymes, so the dosage of quinidine sulfate can be reduced accordingly.
In the case of chiral drugs, deuterium modification can also play a significant role. For example, in the case of thalidomide, the introduction of a deuterated chiral switch (deuterium-enabled chiral switch, DECS) can reduce the formation of the enol intermediate, thereby inhibiting the isomerization of R-thalidomide into the teratogenic S-thalidomide, leading to improved drug safety. Additionally, deuterium modification at the chiral center of a drug can enhance its efficacy or selectivity, with specific examples including DRX-164, PXL065, Y180, and others.
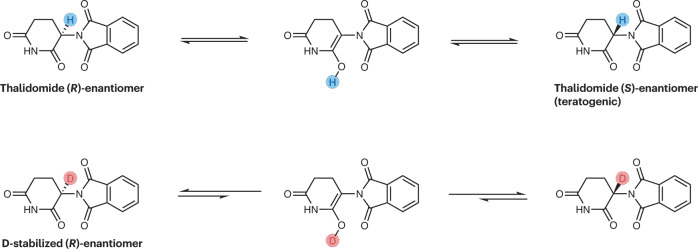
Fig 2. Influence on the interconversion between stereoisomers.[1]
Influence on mechanisms of action: RT001 as a case study
Deuterium modification of drugs can potentially alter their mechanisms of action, leading to changes in their pharmacological activity. ALK001 (d3-vitamin A) is a deuterated form of vitamin A, which can reduce the biosynthesis of vitamin A dimers. Phase II studies for ALK001 are currently underway. RT001 (d2-linoleic acid ethyl ester) is stabilized by deuterium substitution in the molecule, preventing the process of lipid peroxidation. Although the change from H to D is small, it can still influence the interaction between the drug and its target, enhancing the immune response. Deuterium-modified heroin vaccines have been shown to enhance the immune response, demonstrating the potential of deuterium modification to augment the immune response.
Deuterium switches approved so far
Currently, several deuterated drugs have successfully reached the market, and multiple deuterated drugs are in various stages of clinical research. In 2017, the FDA approved the first deuterated drug, deutetrabenazine, which is used to treat Huntington’s disease. In 2021, donafenib was approved for the treatment of various cancers. Donafenib is a deuterated multi-kinase inhibitor that replaces a key metabolic site, pyridyl amine, in the molecule of sorafenib with pyridyl tri-deuteromethyl amine. In head-to-head clinical trials with sorafenib, donafenib showed superior metabolic characteristics, efficacy, and safety. In the same year, VV116, a Remdesivir analog developed using deuterium modification and prodrug strategies, was also approved and launched in Uzbekistan. VV116 has a mechanism of action consistent with Remdesivir but exhibits good oral bioavailability, making it suitable for oral administration.
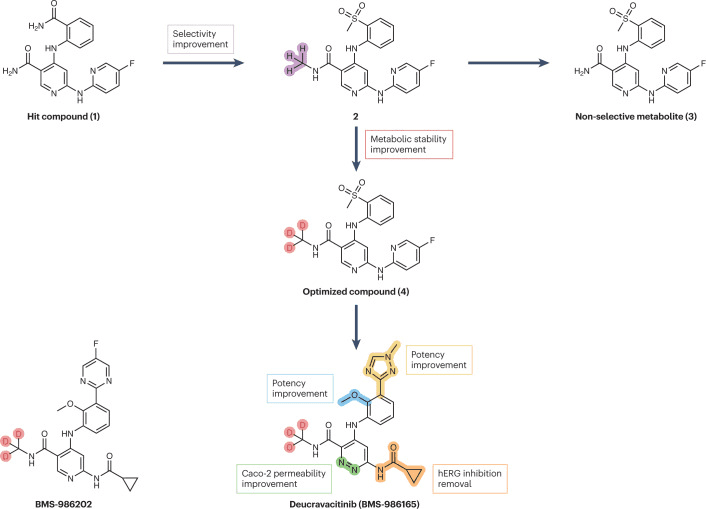
Fig 3. Streamlined representation of the campaigns that led to the development of deucravacitinib and BMS-986202. [1]
It’s worth noting that in September 2022, deucravacitinib was approved for the treatment of psoriasis. This drug represents a new deuterated drug discovered from scratch (deuterium in de novo drug discovery). As shown in the figure above, Bristol-Myers Squibb carried out methyl modifications on the lead compound 1, resulting in compound 2, which exhibited selectivity for TYK2 within the JAK family. However, this compound produced non-selective metabolite 3 during the metabolism process. The introduction of deuterium atoms inhibited N-dealkylation, and compound 4 did not produce non-selective metabolites. Further structural modifications on compound 4 led to the development of deucravacitinib. Bristol-Myers Squibb also has two deuterated compounds, BMS-986322 and BMS-986202, as potential follow-up compounds to deucravacitinib. Additionally, two deuterated molecules designed from scratch, deucrictibant (a bradykinin receptor antagonist) and VX-984, have also entered clinical research.

Fig 4. SWOT analysis of the deuteration approach.[1]
Summary
As mentioned earlier, deuterium (D) and hydrogen (H) have minor differences in atomic size and electronegative characteristics. C-D bonds are more stable than C-H bonds, and deuterium modification can improve a drug’s absorption, distribution, metabolism, excretion, and toxicity (ADMET) in the body. This modification is considered safer than fluorination in terms of electronic properties. However, deuterium modification may also bring some risks, including the cumulative effect of deuterium atoms on drug lipophilicity and the potential for drug-drug interactions (DDI) due to deuterium incorporation.
From a synthetic perspective, introducing a fixed number of deuterium atoms at specific positions and ensuring deuterium efficiency can be challenging. Analyzing isotopic impurities in deuterated active pharmaceutical ingredients (APIs) also presents technical challenges. Moreover, the effects of deuterium modification cannot be easily predicted, and the development of deuterated drugs requires significant financial investment in extensive research. Regulatory agencies require that deuterated drugs demonstrate clear advantages over existing non-deuterated compounds in terms of effectiveness, safety, or dosing frequency. Transitioning from basic research to clinical application for deuterated drugs can be quite challenging.
Reference
- Di Martino, R. M. C., Maxwell, B. D., & Pirali, T. (2023). Deuterium in drug discovery: progress, opportunities and challenges. Nature reviews. Drug discovery, 22(7), 562–584.
- Chandra Mouli, H. M., Vinod, A., Kumari, S., Tiwari, A. K., Kathiravan, M. K., Ravichandiran, V., & Peraman, R. (2023). Deuterated driven new chemical entities: An optimistic way to improve therapeutic efficacy. Bioorganic chemistry, 135, 106490.
- Qian, H. J., Wang, Y., Zhang, M. Q., Xie, Y. C., Wu, Q. Q., Liang, L. Y., Cao, Y., Duan, H. Q., Tian, G. H., Ma, J., Zhang, Z. B., Li, N., Jia, J. Y., Zhang, J., Aisa, H. A., Shen, J. S., Yu, C., Jiang, H. L., Zhang, W. H., Wang, Z., … Liu, G. Y. (2022). Safety, tolerability, and pharmacokinetics of VV116, an oral nucleoside analog against SARS-CoV-2, in Chinese healthy subjects. Acta pharmacologica Sinica, 43(12), 3130–3138.
- Wrobleski, S. T., Moslin, R., Lin, S., Zhang, Y., Spergel, S., Kempson, J., Tokarski, J. S., Strnad, J., Zupa-Fernandez, A., Cheng, L., Shuster, D., Gillooly, K., Yang, X., Heimrich, E., McIntyre, K. W., Chaudhry, C., Khan, J., Ruzanov, M., Tredup, J., Mulligan, D., … Weinstein, D. S. (2019). Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165. Journal of medicinal chemistry, 62(20), 8973–8995.
- Gant T. G. (2014). Using deuterium in drug discovery: leaving the label in the drug. Journal of medicinal chemistry, 57(9), 3595–3611.