Modulating Treg Homeostasis - Improving Cancer Immunotherapy
Abstract
Immunosuppressive regulatory T cells (Tregs) are the main mechanism of tumor immune evasion. Targeting Tregs, especially in the tumor microenvironment (TME), could improve cancer immunotherapy. Research in recent years has revealed the heterogeneity and plasticity of Tregs within tumors, further deepening the complexity of the role of Tregs in tumor immunity and immune response therapy. The phenotypic and functional diversity of Tregs within tumors can influence their response to therapy and may provide new targets for modulating specific Treg subsets
T cell classification
According to the maturation time of T cells, they can be divided into Naive T cells, Effector T cells, and Memory T cells. Based on the types of T cell receptors (TCRs) they express, they can be further categorized into αβ T cells and γδ T cells. According to surface protein characteristics, they can be further classified as CD4+ or CD8+ T cells. Within the CD8+ subset, the main cells are cytotoxic T cells, abbreviated as Tc cells, with a small population of CD8+ Treg cells. Among CD4+ cells, they can be primarily divided into Helper T cells (Th) and Treg cells. Treg cells can be mainly classified into two types: Natural Treg (nTreg), which are generated in the thymus and mediate immunosuppression and tolerance responses through contact or non-contact mechanisms, and Inducible Treg (iTreg), which are induced from Naive T cells and mediate immunosuppression and tolerance responses through contact or non-contact mechanisms.
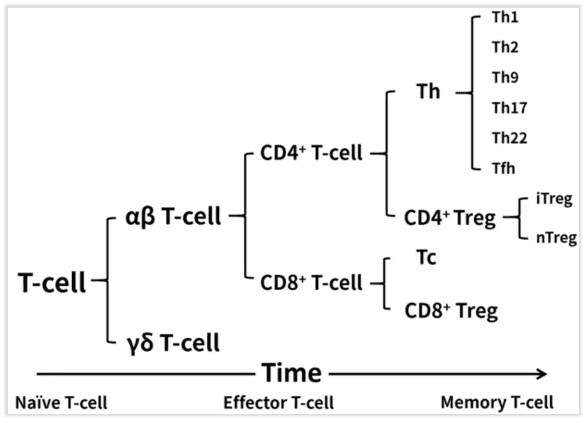
Fig 1. T cell classification
What are Tregs?
Tregs are a type of immunosuppressive CD4+ T cell used to maintain immune tolerance and tissue homeostasis, preventing autoimmunity. In cancer, these mechanisms work together, supporting tumor immune evasion and progression. Growing tumors serve as a source of tissue damage and inflammatory stimulation, to which Tregs typically respond. Therefore, Tregs accumulate and expand in the TME, acting as a feedback mechanism to restore immune and tissue homeostasis, subsequently inhibiting anti-tumor immunity. The elevated frequency of Tregs within the tumor is associated with adverse prognosis in various types of cancer patients, providing a basis for targeting Tregs in cancer immunotherapy.
Increasing evidence suggests that tumor-infiltrating Tregs exhibit a high degree of heterogeneity based on their origin, function, and phenotype (Fig 2). Different TMEs can alter Treg phenotype and stability, enabling them to switch between different functional states. Recent studies have revealed the impact of tumor antigens, co-stimulatory signals, and metabolic products (such as lactate, glucose) on the homeostasis and lineage stability of Tregs within tumors. This indicates that environmental signals may shape the repertoire and plasticity of tumor-infiltrating Tregs through mechanisms involving cellular metabolism and downstream epigenetic recombination, thereby altering the classical Treg transcriptional program coordinated by the Treg-regulating factor Foxp3.
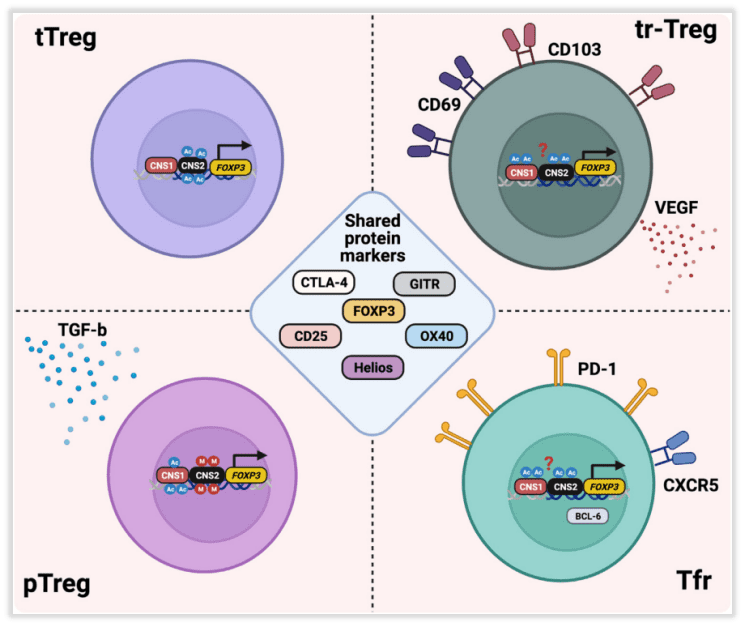
Fig 2. Intratumoral regulatory T cell (Treg) heterogeneity.
Intratumoral Treg subpopulation
Tregs may originate from different lineages and developmental stages, including Tregs derived from thymic selection (tTregs), peripherally induced Tregs (pTregs), and Tregs with specific phenotypes such as tissue-resident Tregs (tr-Tregs) and follicular Tregs (Tfr) (Fig 2).
Early studies suggest that peripherally induced Tregs (pTregs) are the primary source of tumor-associated Tregs, as Tregs are found in tumors, tumor-draining lymph nodes (TDLNs), and even in thymectomized mice. In other studies, it was observed that the function of activated tumor antigen-specific CD4+ T cells in TDLNs is suppressed. This suggests that the accumulation of Tregs within tumors may arise from the conversion of conventional CD4+Foxp3– T cells (Tconv) into pTregs within the TME or TDLNs. However, in several mouse models, there was almost no overlap observed between the Treg and Tconv TCR repertoires, thus negating this hypothesis. In subsequent transplantable lung cancer and melanoma models, it was found that Treg clones were driven by potentially conserved self-antigens, rather than being derived from Tconv conversion. Similar studies have shown the expansion of thymus-dependent, antigen-specific Tregs against prostate self-antigens in mouse prostate tumors, mechanistically demonstrating the presence of tTregs in tumors.
Neuropilin-1 (Nrp1) and Helios (IKZF2) are considered markers to distinguish between tTregs and pTregs, but it is currently unclear whether these factors can truly identify different Treg subpopulations within the TME. More robust markers are needed to differentiate these Treg subpopulations within the tumor microenvironment.
Tr-Treg is a key regulator of TME. In addition to inhibiting T cell function, Tr-Treg can also play an immune-independent tumor promotion role, such as through two regulatory proteins (CD69 and CD103) and vascular endothelial growth factor (VEGF). The production of Tregs induces epithelial tumor cell proliferation and neovascularization, and VEGF production by Tregs increases during hypoxia, a common feature of TME. These functions of Tr-Tregs may have an important impact in determining the outcome of immunotherapy. Therefore, it is crucial to deepen the characterization of Tr-Treg in tumor development, progression, and treatment response.
Tfr cells represent another subset of Tregs within tumors. They are found in various types of cancers, including non-small cell lung cancer, where they are primarily located in tertiary lymphoid structures (TLSs). They inhibit the highest expression of CTLA-4 and programmed cell death protein 1 (PD-1) in tumor-infiltrating Tregs. There is a high degree of TCR sharing between Tfr and Tregs, suggesting that Tregs may transform into Tfr in an antigen-dependent manner. Mechanistic studies in mice indicate that Tfr cells possess superior inhibitory capabilities compared to Tregs and exhibit a highly responsive reaction to anti-PD-1 therapy. Tfr cells expand after anti-PD-1 treatment and suppress anti-tumor activity.
Intratumoral TCR signal intensity affects Treg stability
TCR signaling is crucial for the development and functional integrity of Tregs. Compared to conventional CD4+Foxp3– (Tconv) cells, Tregs exhibit tolerance to high-intensity TCR signals during their development. In fact, TCR signaling is essential for the induction of Foxp3, as the loss of major histocompatibility complex II (MHC-II) primarily results in the loss of Foxp3+ thymic cells in mice. In the periphery, brief high-intensity TCR signals can induce the development of pTregs.
Similar to Tconv cells, TCR activation in Tregs leads to protein kinase signaling cascades, activating key transcription factors (Fig 3). Specifically, the CARD11-BCL10-MALT1 (CBM) signalosome links TCR signaling to nuclear factor κB (NF-κB) activation, which is crucial for Treg development and function. Mice with CBM defects exhibit severe deficiencies in Tregs, primarily dependent on the loss of MALT1 proteinase activity. Recent studies have reported that mice treated with the MALT1 proteinase inhibitor, ibrutinib, demonstrate better control over tumor growth and reduced tumor-infiltrating Tregs. In addition to reducing Treg immunosuppression, MALT1 inhibitors induce fragility in tumor-infiltrating Tregs (IFN-γ production), thereby supporting anti-tumor responses. It’s worth noting that the CBM complex and MALT1 activity promote the upregulation of CTLA-4 in Tregs upon TCR activation, which underlies the fundamental principle of dual targeting with MALT1 inhibition and immune checkpoint blockade (ICB) therapy.
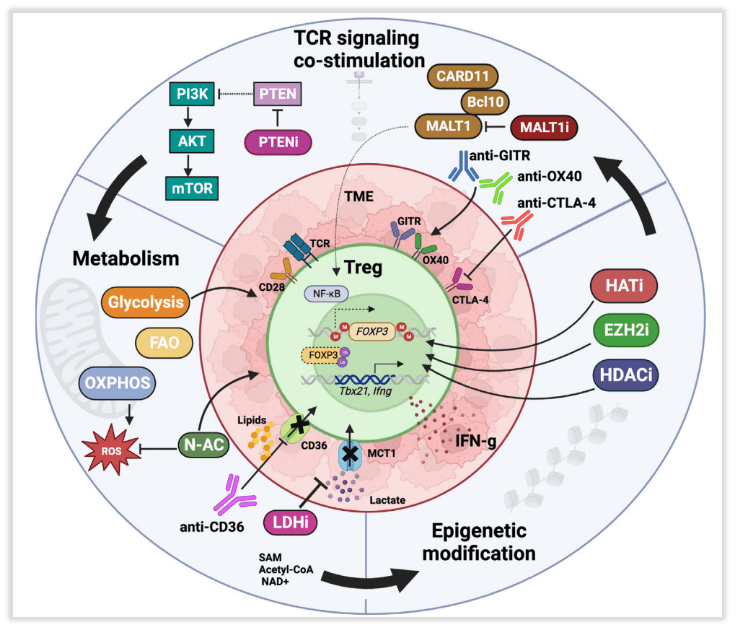
Fig 3. Molecular and related pharmacological interventions leading to instability of regulatory T cells (Tregs)
Tumor-infiltrating Tregs primarily express proteins indicative of antigen recognition and TCR activation. Their function involves downregulating naive T cell markers (such as CD62L, CD45RA), increasing Foxp3 expression, and upregulating co-stimulatory and inhibitory receptors, including glucocorticoid-induced tumor necrosis factor-related protein (GITR), OX40, CD25, CTLA-4, and PD-1. These phenotypes likely sustain signal transduction for Treg survival and inhibitory function. In mouse models, targeting OX40, GITR, and CTLA-4 preferentially modulates tumor-infiltrating Tregs, although this has not been conclusively demonstrated in humans. It has been reported that the use of GITR agonist antibodies can regulate the phenotype of both tumor-infiltrating and peripheral Tregs with effective phenotypes. GITR or OX40 agonists reduce the phenotype stability and inhibitory function of human and mouse Tregs, possibly due to overstimulation of Tregs leading to reduced survival rates.
The metabolism and stress response of intratumoral Treg integrity
Tregs exhibit characteristic metabolic preferences, which are induced by the integration of TCR and co-stimulatory signaling pathways and are utilized to maintain Treg immunosuppression. The specific oxidative metabolism of Tregs complements the oxidative metabolism of tumor cells, which often rely on glycolysis. Compared to effector immune cells that also rely on glycolysis and compete for glucose with tumor cells, Tregs in the TME have a metabolic advantage. The fine-tuning of TCR signaling and co-stimulatory pathways in Tregs supports their metabolism, stability, and functionality. Therefore, nutritional changes in the TME can significantly influence the phenotypic, metabolic, and functional characteristics of tumor-infiltrating Tregs, as described below.
1. Treg metabolic vulnerability in the TME
Tumor-infiltrating Tregs heavily rely on lipid oxidation, a fact demonstrated by the specific loss of the fatty acid scavenger receptor CD36. Tregs deficient in CD36 exhibit alterations in fatty acid metabolism and mitochondrial adaptability, resulting in increased production of IFN-γ and tumor necrosis factor alpha (TNF-α), along with reduced tumor infiltration. CD36 represents a promising target for modulating tumor-infiltrating Tregs while limiting systemic toxicity. Building on this, the use of CD36-blocking antibodies in mouse models achieved regression of invasive, spontaneous melanomas. Similar metabolic targets include Monocarboxylate Transporter 1 (MCT1) and Lactate Dehydrogenase (LDH) involved in lactate metabolism.
Enhancing Treg glucose metabolism can also reduce Treg’s suppressive function and enhance anti-tumor immunity. Early studies indicate that overexpressing the glucose transporter Glut1 in Tregs shifts their metabolism from oxidation to glycolysis, reducing the phenotypic and functional stability of Tregs in vitro and in vivo. CTLA-4 has been reported to support Treg oxidative metabolism by blocking CD28-mediated glycolysis induction. CTLA-4 blockade induces Treg glucose metabolism and fragility, thereby improving therapeutic efficacy.
2. The Regulation of Tumor-Infiltrating Treg Stability by the Hypoxic Response
The low oxygen tension (hypoxia) in the TME affects the function and stability of Tregs. In inflammatory tissues, it has been found that hypoxia-inducible factor-1 (HIF-1)α subunit binds to the FOXP3 promoter, enhancing FOXP3 transcription in Tregs, which is necessary for their survival and function. Therefore, Tregs with a deficiency in HIF-1α lose their suppressive ability, indicating the importance of HIF-1α for Treg function. However, studies have found that excessive HIF-1α activity in Tregs can actually inhibit their activity and promote inflammation. HIF-1α can bind to the Foxp3 protein, leading to its degradation. Additionally, HIF-1α can re-polarize Tregs into Th17 cells by upregulating the Th17 master regulator, retinoic acid receptor (RAR)-related orphan receptor (ROR)γt, while weakening Treg development. These HIF-1α-regulated mechanisms, in response to changes in inflammation and hypoxia within the TME, may thus modulate the function and stability of Tregs.
Summary
The differentiation of Tregs arises from the integration of environmental signals, including antigens, co-stimulatory ligands, and cytokines. These signals converge onto specific epigenetic programs activating classic Treg genes, including Foxp3. The stability of Tregs also depends on the utilization of specific metabolic pathways to generate energy. Because metabolic intermediates alter the function of epigenetic enzymes, Treg metabolic preferences may contribute to strengthening Treg epigenetic programs. These findings suggest subtle interactions between intracellular metabolic products and epigenetic enzymes controlled by immune receptor signaling in determining Treg phenotype, stability, and function. These mechanisms may lead to the heterogeneity of Tregs within tumors.
Precise regulation of these pathways enables us to target the Treg subsets that most effectively promote tumors, thus helping to realize the full potential of cancer immunotherapy. In this direction, the use of next-generation single-cell and spatial sequencing methods enhances our capacity to address the heterogeneity of tumor-infiltrating Tregs, allowing us to understand the mechanisms underlying the accumulation of different Treg subsets in tumors and their relative contributions to tumor progression. Targeting their antigen targets helps reduce the inhibitory Treg subsets in tumor immunity while maintaining peripheral tolerance, thereby enhancing the effectiveness of cancer immunotherapy.