KRAS-Targeted Drugs: From Discovery to Clinical Application
Abstract
KRAS has long been considered an elusive target in oncology due to its structural resistance to conventional drug design. However, recent breakthroughs—especially the discovery of the KRAS G12C mutation—have enabled the development of covalent inhibitors that lock the protein in its inactive GDP-bound state. The FDA approvals of Sotorasib and Adagrasib have transformed the treatment landscape for KRAS-mutant non-small cell lung cancer (NSCLC) and colorectal cancer (CRC), validating KRAS as a druggable oncogene. This advancement has triggered a new wave of investigational therapies, including Garsorasib, D3S-001, and Glecirasib, which aim to improve efficacy and address resistance mechanisms. Despite early success, tumor adaptation through secondary mutations, bypass pathway activation, and phenotypic changes remains a significant barrier. To overcome this, researchers are exploring synergistic combination therapies involving immune checkpoint inhibitors, SHP2 inhibitors, and EGFR blockade. Continued innovation and personalized strategies are critical to sustaining therapeutic benefits and expanding KRAS-targeted therapy to a broader spectrum of cancers. This review summarizes the journey of KRAS-targeted drugs from discovery to clinical application, emphasizing current challenges, mechanisms of action, resistance pathways, and future directions in this rapidly evolving field.
Introduction: What Is KRAS and Why Was It ‘Undruggable’?
KRAS (Kirsten rat sarcoma viral oncogene homolog) is a small GTPase and a member of the RAS family of proteins, playing a critical role in regulating cell proliferation, differentiation, and survival. Under physiological conditions, KRAS alternates between an active GTP-bound state and an inactive GDP-bound state, acting as a molecular switch for multiple signaling pathways, including the MAPK and PI3K cascades. However, mutations in the KRAS gene—especially at codon 12 (e.g., G12C, G12D, G12V)—disrupt its GTPase activity, resulting in persistent activation that drives uncontrolled cell division and cancer progression.
KRAS mutations are among the most prevalent oncogenic drivers in human cancers. They are found in approximately 30% of non-small cell lung cancer (NSCLC) cases, 40% of colorectal cancers, and over 90% of pancreatic ductal adenocarcinomas. Despite their prevalence, efforts to develop drugs targeting KRAS were unsuccessful for decades. This was due to the protein’s high affinity for GTP/GDP and the absence of deep binding pockets on its surface—earning it the label “undruggable.”

The narrative began to change with the discovery of the KRAS G12C mutation and the realization that this specific alteration introduced a reactive cysteine residue amenable to covalent inhibition. This breakthrough enabled the design of small molecules that could irreversibly bind to the mutant KRAS in its inactive (GDP-bound) state, shutting down its signaling capacity. These advances marked the beginning of a new era in precision oncology, transforming KRAS from a frustrating drug target into one of the most promising domains in targeted cancer therapy.
Breakthrough in Targeting KRAS
For decades, KRAS was a symbol of drug development failure—a high-priority oncogene deemed therapeutically unreachable. The tide began to shift with the identification of the KRAS G12C mutation, a specific alteration that replaces glycine with cysteine at codon 12. This mutation, found in about 13% of NSCLC and 3% of colorectal cancers, introduced a new therapeutic vulnerability. The cysteine residue enabled researchers to design covalent inhibitors that selectively and irreversibly bind to the mutant KRAS protein in its GDP-bound (inactive) state.
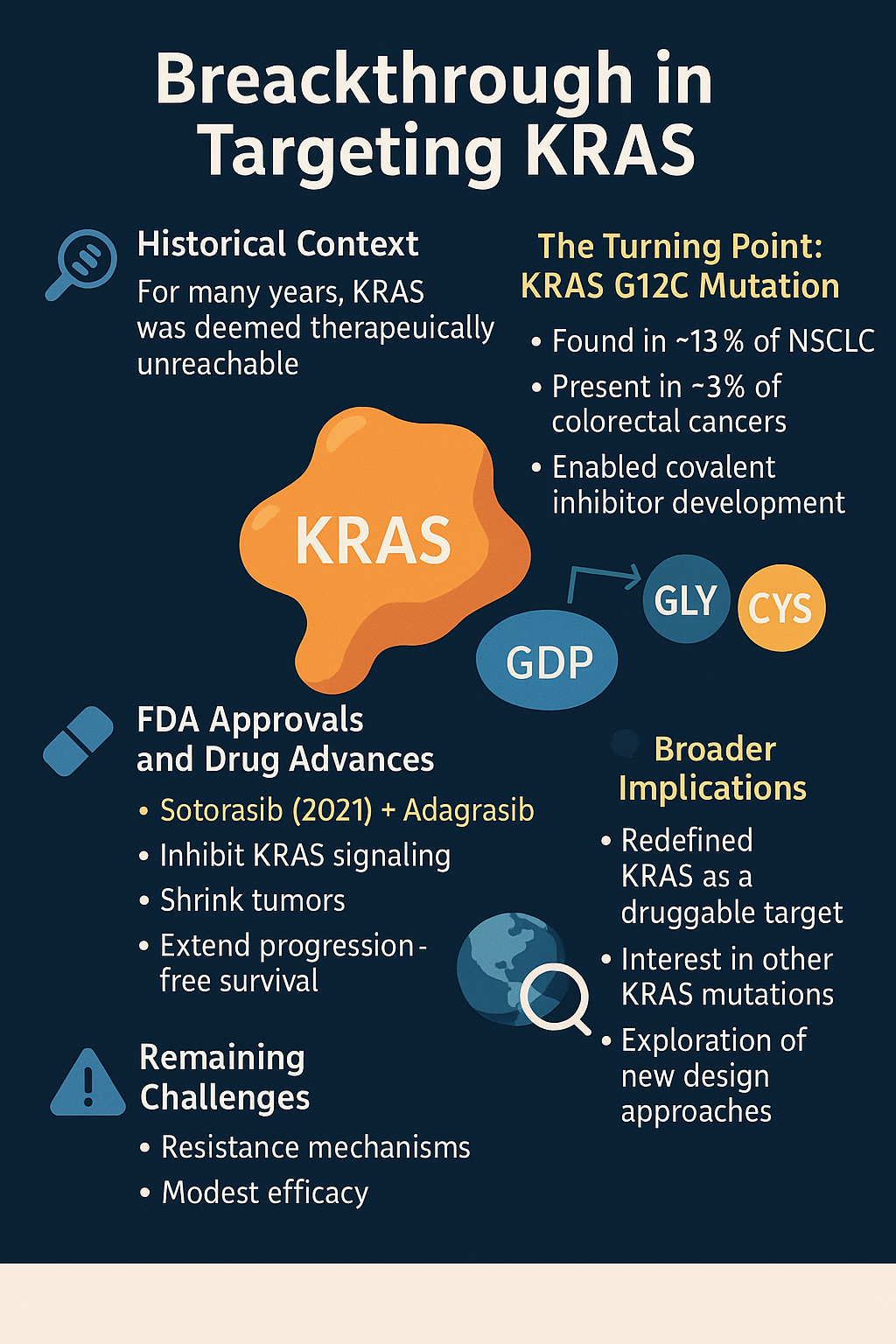
In 2021, the U.S. FDA approved Sotorasib (AMG 510)—the first targeted KRAS inhibitor—for advanced NSCLC with KRAS G12C mutations. This approval marked a monumental breakthrough in oncology, representing decades of molecular and structural biology advances. Soon after, Adagrasib (MRTX849) followed, showing promise in both NSCLC and colorectal cancer. These drugs demonstrated the ability to inhibit KRAS signaling, shrink tumors, and extend progression-free survival.
The success of G12C inhibitors has redefined KRAS as a druggable target, sparking intense pharmaceutical competition and academic research into other KRAS variants like G12D and G12V. Additionally, it has encouraged broader exploration of structure-guided design, allosteric inhibition, and covalent chemistry. While challenges remain—including resistance and modest efficacy in certain tumor types—the era of targeting KRAS has indisputably begun.
Approved KRAS Inhibitors
The clinical validation of KRAS-targeted therapy came with the approvals of Sotorasib (AMG 510) and Adagrasib (MRTX849), two selective KRAS G12C inhibitors that made history in precision oncology. Both agents work by covalently binding to the cysteine introduced by the G12C mutation, irreversibly locking KRAS in its inactive state and disrupting downstream oncogenic signaling.
Sotorasib, the first to market, was approved by the FDA in 2021 for KRAS G12C-mutated NSCLC. In the CodeBreaK 100 trial, it demonstrated a 32% objective response rate (ORR) and a 6.3-month median progression-free survival (PFS). Patients with prior platinum-based chemotherapy and immunotherapy experienced notable tumor shrinkage with manageable toxicity.
Adagrasib followed in 2022 with approval for NSCLC and broader evaluations in colorectal cancer (CRC). The KRYSTAL-1 trial reported an ORR of 43% in NSCLC and 22% in CRC, highlighting its ability to penetrate the central nervous system and showing potential synergy in combination regimens with EGFR inhibitors like cetuximab.
These approvals were not only clinical milestones but also proof-of-concept achievements that transformed KRAS from an elusive target to a new standard in molecular oncology. While these inhibitors primarily address G12C mutations, their success has galvanized the race to develop agents for other KRAS variants, such as G12D, G12V, and Q61H. Their journey from bench to bedside continues to serve as a model for next-generation drug discovery.
Emerging KRAS Inhibitors in Clinical Trials
Following the success of Sotorasib and Adagrasib, the field of KRAS-targeted therapy is expanding rapidly, with multiple new inhibitors entering clinical trials. These second-wave agents aim to improve efficacy, tackle central nervous system (CNS) metastases, and overcome resistance mechanisms observed with first-generation drugs.
One of the most prominent emerging inhibitors is Garsorasib (D-1553), which has shown strong preclinical activity and is undergoing phase II trials in China and internationally. In NSCLC patients harboring KRAS G12C mutations, Garsorasib has demonstrated an objective response rate (ORR) of 34.8% and promising CNS activity. Similarly, D3S-001 is in phase I trials, exhibiting tolerability and potential in solid tumors, including colorectal and pancreatic cancers.
Other notable candidates include Glecirasib (JAB-21822) and IBI351, both of which are selective G12C inhibitors being tested in China for advanced solid tumors. These drugs aim to offer differentiated pharmacokinetic profiles and broader tissue penetration, including the brain. Their development also reflects growing innovation outside Western pharmaceutical pipelines.
These investigational therapies are being studied both as monotherapies and in combination regimens, especially with immune checkpoint inhibitors, SHP2 inhibitors, and EGFR inhibitors. The goal is to improve duration of response and combat early resistance. While results remain preliminary, these emerging agents are crucial to establishing a robust and diverse KRAS-targeted treatment ecosystem that can serve a wider patient population.
Mechanisms of Action
KRAS inhibitors are structurally designed to exploit a specific vulnerability introduced by the G12C mutation, which substitutes glycine with cysteine. This cysteine provides a reactive nucleophilic target for covalent binding. The inhibitors, such as Sotorasib and Adagrasib, preferentially bind to KRAS in its inactive GDP-bound state, effectively locking the protein in a non-functional configuration.
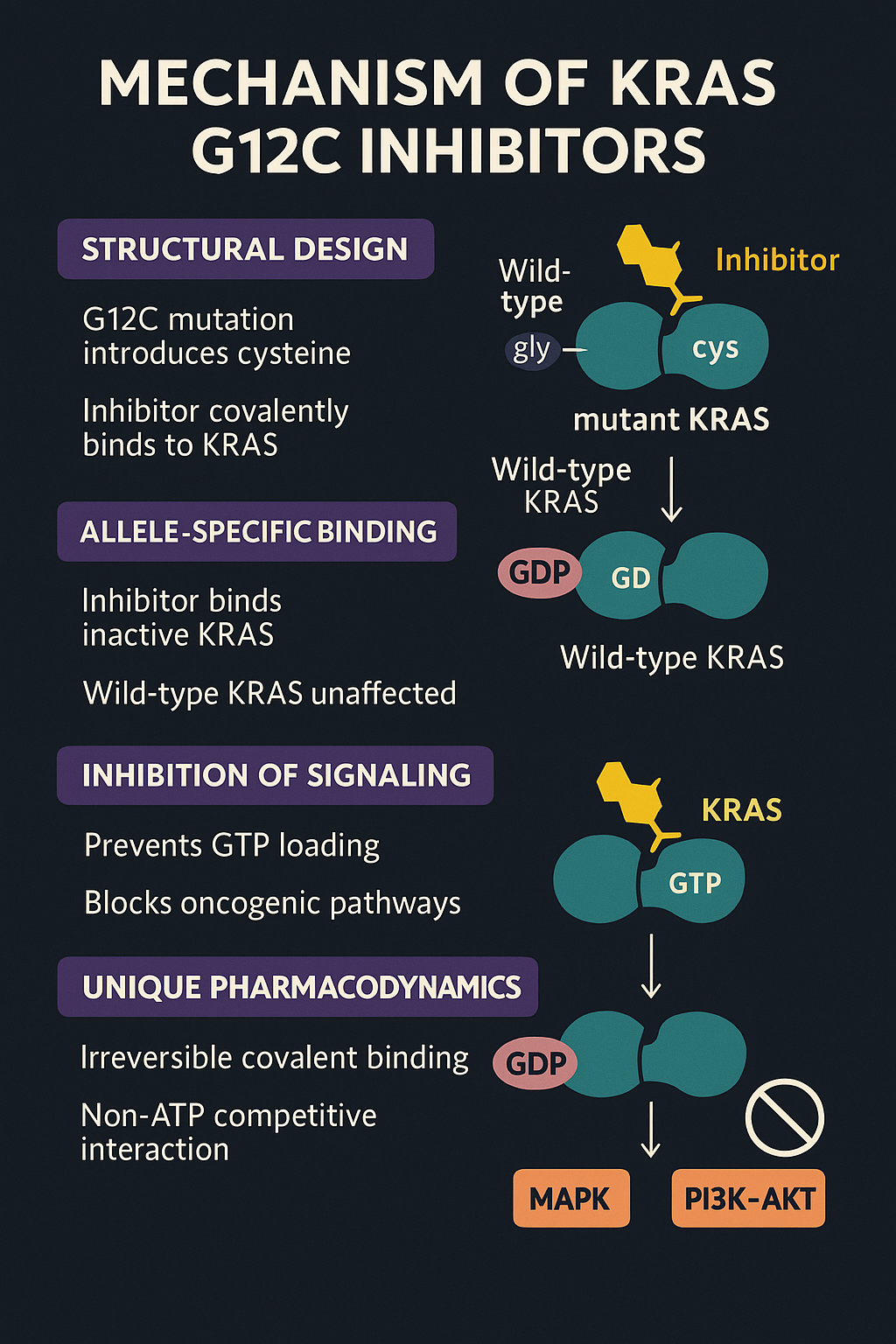
The innovation behind this approach lies in disrupting the KRAS signaling cycle, where the protein toggles between active (GTP-bound) and inactive (GDP-bound) forms. Mutant KRAS typically remains in a constitutively active state, promoting cell proliferation through downstream pathways like MAPK and PI3K–AKT. By irreversibly binding to KRAS G12C, these drugs prevent GTP loading and halt downstream oncogenic signaling.
Notably, these inhibitors are allele-specific, meaning they do not affect wild-type KRAS or other RAS isoforms—thereby improving therapeutic selectivity and reducing systemic toxicity. This mechanism is fundamentally different from traditional kinase inhibitors and represents a non-ATP competitive, non-reversible interaction, offering a unique pharmacodynamic profile.
The specificity of action also means that drugs like Sotorasib are ineffective against non-G12C variants. As such, new agents are being developed to target other mutations like G12D, G12V, and Q61H, using alternative strategies such as allosteric modulation, switch II pocket targeting, and pan-KRAS inhibition.
Understanding this mechanism is vital not only for clinical application but also for the rational design of combination therapies that enhance efficacy and delay resistance.
Resistance to KRAS Inhibitors
Despite their initial promise, KRAS G12C inhibitors like Sotorasib and Adagrasib often encounter resistance, limiting their long-term effectiveness. Resistance mechanisms can be either intrinsic (present before treatment) or acquired (developed during therapy), and they often emerge within months of treatment initiation.
A primary mode of resistance involves secondary mutations in the KRAS gene itself—such as at residues G13, Q61, or Y96—which alter the binding pocket and prevent covalent inhibition. These secondary KRAS mutations can confer cross-resistance to both Sotorasib and Adagrasib.
In addition, many tumors activate bypass signaling pathways. For example, EGFR and MET amplification, FGFR activation, or upregulation of wild-type RAS isoforms (e.g., NRAS, HRAS) can reactivate the downstream MAPK pathway, undermining the inhibitor’s effect. This phenomenon is especially pronounced in colorectal cancer, where EGFR-mediated feedback is common.
Phenotypic changes in tumor cells also contribute to resistance. These include epithelial-to-mesenchymal transition (EMT), alterations in tumor microenvironment, and histological transformation to a different cancer subtype.
Overcoming resistance requires an evolving therapeutic approach. Identifying biomarkers of early resistance, performing liquid biopsies to monitor KRAS variants, and employing combination regimens are current strategies under investigation.
Combination Strategies
To address the limited durability of KRAS G12C inhibitors as monotherapies, researchers are increasingly exploring combination strategies. These approaches aim to enhance efficacy, prolong response duration, and delay or overcome resistance.
One prominent strategy is combining KRAS inhibitors with immune checkpoint inhibitors such as anti-PD-1/PD-L1 antibodies. Preclinical studies suggest that KRAS inhibition may promote a more immunogenic tumor microenvironment, rendering cancer cells more susceptible to immune-mediated killing.
Another key focus is the co-targeting of upstream RTK pathways, especially EGFR and MET, which frequently reactivate downstream signaling even during KRAS inhibition. The combination of Adagrasib with Cetuximab (an EGFR antibody) in colorectal cancer patients has shown improved response rates in early-phase trials.
SHP2 inhibitors, which block signaling from various receptor tyrosine kinases to RAS, are also under evaluation. By impeding upstream activation, these agents can synergize with KRAS inhibitors and prevent reactivation of the MAPK pathway. Similarly, combining with MEK inhibitors, which act downstream, represents a vertical targeting strategy to suppress escape mechanisms.
Other exploratory combinations involve agents like CDK4/6 inhibitors, PI3K inhibitors, and even chemotherapy in select settings. The key to these strategies lies in understanding tumor heterogeneity and tailoring treatment to specific escape mechanisms observed in different tumor types.
While combination therapies increase complexity and potential toxicity, they represent the most promising avenue to maximize the therapeutic potential of KRAS inhibitors and move toward long-term disease control.
Conclusion
The evolution of KRAS-targeted therapies represents one of the most significant breakthroughs in precision oncology. Once considered an “undruggable” target due to its smooth protein surface and high affinity for GTP/GDP, KRAS is now at the center of innovative cancer treatment strategies. The successful development and approval of KRAS G12C inhibitors such as Sotorasib and Adagrasib not only demonstrated therapeutic viability but also reignited global interest in targeting other KRAS mutations like G12D and G12V.
Despite initial success, challenges remain—particularly resistance mechanisms that limit long-term efficacy. This has spurred a wave of combination strategies aimed at enhancing durability and circumventing tumor escape. The emergence of next-generation inhibitors such as Garsorasib and Glecirasib, along with novel agents targeting broader KRAS mutations or exploiting upstream/downstream pathways, reflects a vibrant and fast-growing research landscape.
As understanding deepens around the biology of KRAS-driven cancers and how they adapt, the future lies in personalized regimens informed by genetic profiling, resistance tracking, and rational drug combinations. Ultimately, the KRAS story is one of resilience and innovation—transforming an intractable target into a symbol of what’s possible in modern oncology. Continued progress could redefine therapeutic standards across several of the most lethal cancer types.
References
Long SA, Amparo AM, Goodhart G, Ahmad SA, Waters AM. Evaluation of KRAS inhibitor-directed therapies for pancreatic cancer treatment. Front Oncol. 2024 May 10;14:1402128. doi: 10.3389/fonc.2024.1402128. PMID: 38800401; PMCID: PMC11116577.
https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2024.1402128/full
Zhao X, Zheng Y, Wang Y, Zhang M, Dong Z, Liu Y, Sun M. The Potential Treatment Options and Combination Strategies of KRAS-Mutated Lung Cancer. Onco Targets Ther. 2024 Nov 15;17:1041-1057. doi: 10.2147/OTT.S484209. PMID: 39564454; PMCID: PMC11575457.
https://www.tandfonline.com/doi/full/10.2147/OTT.S484209
Pandey D, Chauhan SC, Kashyap VK, Roy KK. Structural insights into small-molecule KRAS inhibitors for targeting KRAS mutant cancers. Eur J Med Chem. 2024 Nov 5;277:116771. doi: 10.1016/j.ejmech.2024.116771. Epub 2024 Aug 15. PMID: 39167893.
https://www.sciencedirect.com/science/article/abs/pii/S0223523424006524
Li Z, Zhou Z, Zhang N, Tian B, Chen X, Zhao H, Wang H. Hepatitis associated with immune checkpoint inhibitors-based combinations of other therapies: A real-world pharmacovigilance analysis based on the FDA adverse event reporting system (FAERS) database. Cancer Immunol Immunother. 2024 Nov 15;74(1):25. doi: 10.1007/s00262-024-03858-4. PMID: 39546001; PMCID: PMC11568091.
https://link.springer.com/article/10.1007/s00262-024-03858-4
Pandey D, Roy KK. Decoding KRAS dynamics: Exploring the impact of mutations and inhibitor binding. Arch Biochem Biophys. 2025 Feb;764:110279. doi: 10.1016/j.abb.2024.110279. Epub 2024 Dec 20. PMID: 39710177.
https://www.sciencedirect.com/science/article/abs/pii/S0003986124004016