How Lipoprotein Lipase Could Revolutionize Heart Disease Treatment in Metabolic Syndrome
Abstract
Lipoprotein lipase (LPL) is increasingly recognized as a pivotal enzyme in the pathophysiology of cardiovascular disease, particularly in individuals with obesity and type 2 diabetes. Traditionally viewed as a facilitator of fatty acid delivery for energy metabolism, LPL also plays a significant role in atherogenesis when misregulated in immune cells. This blog explores LPL’s dual functionality—supporting cardiac energetics while contributing to vascular plaque formation—and discusses the emerging landscape of LPL-targeted therapies. By highlighting tissue-specific regulation and the therapeutic potential of incretins, APOC3 and ANGPTL inhibitors, and omega-3 fatty acids, we argue that precision modulation of LPL could redefine strategies for preventing diabetic cardiomyopathy and atherosclerosis. As cardiovascular complications continue to drive morbidity in metabolic disorders, LPL stands out as a novel and promising intervention point.
The Overlooked Link Between Diabetes, Obesity, and Heart Disease
In the 21st century, few health challenges have surged with as much intensity and complexity as the twin epidemics of obesity and type 2 diabetes. As waistlines expand and insulin resistance becomes increasingly common, a third silent threat looms larger than ever: cardiovascular disease (CVD). Despite significant advances in glucose-lowering medications and lifestyle interventions, cardiovascular complications remain the leading cause of mortality in people with metabolic disorders.
Traditionally, clinical care for diabetes has focused on controlling blood glucose levels and reducing body weight. While these remain essential pillars of disease management, they do not adequately address the metabolic changes in the heart that often precede or exacerbate heart failure. In fact, many patients develop diabetic cardiomyopathy, a form of heart disease independent of hypertension or coronary artery blockages, driven largely by disrupted fuel metabolism within heart tissue.
One of the central shifts that occurs in the diabetic heart is a preference for fatty acid (FA) metabolism over glucose. Although this adaptation initially helps meet the heart’s high energy demands, chronic reliance on FA oxidation results in lipotoxicity, mitochondrial stress, and eventually, impaired cardiac function. Recent research is turning its attention to the enzyme Lipoprotein Lipase (LPL)—a gatekeeper of fatty acid delivery—as a potential culprit and therapeutic target.
LPL is responsible for hydrolyzing triglycerides in circulating lipoproteins, releasing free fatty acids that the heart and other tissues use for energy. Its misregulation in diabetes not only disrupts cardiac metabolism but also contributes to atherosclerotic plaque formation via its action in macrophages. This dual role—beneficial in the heart but harmful in the vasculature—makes LPL an especially intriguing target for future treatments.
As therapeutic options that specifically target cardiac lipid metabolism are scarce, understanding how LPL functions across tissues could pave the way for precision therapies that reduce cardiovascular risk in people with obesity and diabetes. It’s a promising new frontier in cardiometabolic health—one where lipid biology meets heart protection.
What Is Lipoprotein Lipase and Why It Matters
At the heart of lipid metabolism lies a critical enzyme known as Lipoprotein Lipase (LPL). This enzyme plays a foundational role in energy distribution by catalyzing the hydrolysis of triglycerides (TG) in circulating chylomicrons and very-low-density lipoproteins (VLDL) into free fatty acids (FAs) and glycerol. The released FAs are then taken up by tissues like the heart, skeletal muscle, and adipose tissue, where they are either used for energy production or stored for future use.
LPL is produced primarily in parenchymal cells, such as cardiomyocytes in the heart, and then transported to the luminal surface of capillary endothelial cells with the help of GPIHBP1, a key anchoring protein. This positioning allows LPL to act on lipoproteins in the bloodstream, making it a gatekeeper for tissue-specific FA uptake.
In the heart, LPL is essential for supplying the fatty acids required for mitochondrial oxidation, which fuels the constant, rhythmic contractions of cardiac muscle. Under healthy conditions, the heart derives about 70% of its energy from FA oxidation, with glucose and other substrates contributing the rest. However, this balance is disrupted in diabetic conditions, where LPL activity is altered. In early stages of diabetes, coronary LPL activity increases, helping to meet the energy demand. But in severe or prolonged diabetes, LPL activity declines, pushing the heart toward reliance on non-esterified fatty acids (NEFA)—a shift that can cause lipid accumulation, mitochondrial stress, and contractile dysfunction.
Interestingly, LPL is not just a metabolic enzyme. In macrophages, LPL also acts as a bridging molecule, promoting the uptake of remnant lipoproteins and fostering foam cell formation—a hallmark of atherosclerosis. This dual role of LPL—vital in the heart, yet potentially harmful in vascular immune cells—underscores the importance of contextual and tissue-specific regulation.
Because LPL influences both lipid clearance and energy substrate availability, disruptions in its function resonate across multiple systems. Understanding how LPL behaves in different cellular environments may be the key to developing targeted therapies for metabolic heart disease and atherosclerosis alike.
The Dual Nature of LPL—Hero in the Heart, Villain in the Arteries
Lipoprotein lipase (LPL) is a molecular paradox: while it is essential for maintaining cardiac energy balance, it can also contribute to vascular damage and plaque buildup. This duality underscores the complexity of targeting LPL therapeutically, especially in individuals with diabetes or obesity, where both heart and vascular health are compromised.
In the heart, LPL plays a beneficial role by facilitating the hydrolysis of triglyceride-rich lipoproteins and enabling the delivery of fatty acids (FAs) to cardiomyocytes. These FAs are then oxidized in mitochondria to meet the high energy demands of cardiac muscle. In moderate stages of diabetes, increased LPL activity helps sustain energy production through FA oxidation. However, in advanced diabetes, LPL activity declines, and the heart becomes reliant on circulating non-esterified fatty acids (NEFA). This shift contributes to lipotoxicity, mitochondrial overload, and ultimately, heart failure.
Paradoxically, the same enzyme that fuels the heart can also fuel atherosclerosis when expressed in the wrong cell types. In macrophages, LPL acts as a bridging molecule, helping these immune cells take up remnant lipoprotein particles. This leads to the transformation of macrophages into foam cells, a key event in the formation of atherosclerotic plaques. Studies in animal models have shown that while global overexpression of LPL can be protective, macrophage-specific LPL overexpression actually accelerates plaque formation.
Furthermore, smooth muscle cells (SMCs) in human atherosclerotic lesions have also been found to express LPL. These cells, once contractile, shift to a synthetic and phagocytic phenotype under atherogenic conditions, enhancing their ability to internalize lipids. The presence of LPL in SMCs may facilitate this transformation, contributing to plaque complexity and vulnerability.
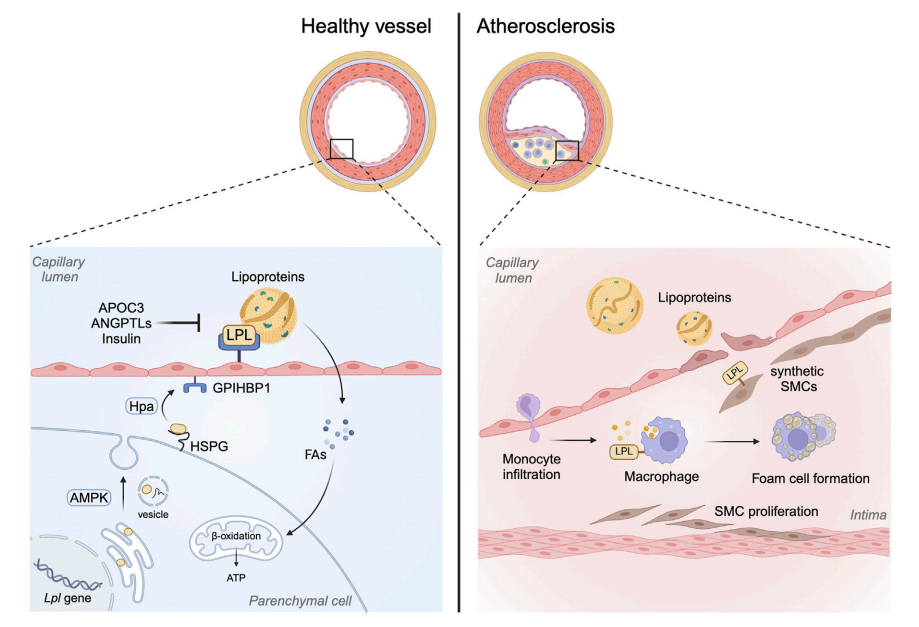
FIGURE 1. Role of LPL beyond its lipolytic action
These tissue-specific actions of LPL highlight a central challenge: any therapeutic strategy involving LPL must distinguish between its cardioprotective and pro-atherogenic effects. Precision targeting—boosting LPL in the heart while suppressing it in macrophages—may be the key to harnessing its full therapeutic potential.
Targeting LPL – Emerging Therapies and Research
As the scientific community continues to explore the metabolic roots of cardiovascular disease, Lipoprotein Lipase (LPL) is emerging as a compelling therapeutic target. Because LPL plays a central role in lipid clearance, fatty acid delivery, and energy metabolism, modulating its activity could offer new strategies for managing diabetic cardiomyopathy and atherosclerosis—two major complications of obesity and type 2 diabetes.
Several drug classes and experimental therapies are now being investigated for their ability to regulate LPL either directly or indirectly. One of the most promising areas involves incretin-based therapies, particularly glucagon-like peptide-1 (GLP-1) receptor agonists and glucose-dependent insulinotropic polypeptide (GIP) analogs. While GLP-1 therapies such as semaglutide are already known to improve glycemic control and reduce cardiovascular risk, their impact on lipid metabolism is still being unraveled. Studies suggest GLP-1 may influence lipoprotein secretion and cholesterol handling, whereas GIP has been shown to stimulate LPL activity, especially in adipose tissue, where it promotes triglyceride clearance and fat storage.
Beyond incretins, LPL regulators such as Apolipoprotein C3 (APOC3) and Angiopoietin-like proteins (ANGPTL3 and ANGPTL4) are gaining attention. Both APOC3 and ANGPTLs act as endogenous inhibitors of LPL, blocking its ability to hydrolyze triglycerides. Therapeutics targeting these molecules—such as volanesorsen and evinacumab—aim to lower triglyceride levels and improve lipid partitioning, potentially reducing cardiovascular events in high-risk patients.
Other agents like omega-3 fatty acids (e.g., icosapent ethyl) and fibrates also modulate LPL through gene expression, enzyme stabilization, or indirect lipid signaling. These drugs not only enhance LPL-mediated triglyceride clearance but may also reduce inflammation and oxidative stress, especially in the context of atherosclerosis.
However, a major therapeutic challenge remains: LPL behaves differently in different tissues. While boosting LPL in the heart may protect against energy deficiency and lipotoxicity, doing the same in macrophages or smooth muscle cells could exacerbate plaque formation. Therefore, cell-specific modulation—enhancing LPL in the myocardium while suppressing it in immune cells—may offer a more refined and safer approach to treatment.
The Future of Heart Disease Prevention – Why LPL Is the Next Big Thing
As rates of obesity and type 2 diabetes continue to rise globally, so too does the burden of cardiovascular disease (CVD). Despite widespread use of glucose-lowering medications and statins, many individuals with metabolic syndrome remain at high residual risk for heart failure and atherosclerotic events. This disconnect has prompted researchers to look beyond traditional targets. One molecule stands out in this evolving landscape: Lipoprotein Lipase (LPL).
LPL’s central role in fatty acid metabolism, triglyceride clearance, and tissue-specific energy balance positions it uniquely at the crossroads of metabolism and cardiovascular health. In the heart, LPL ensures a steady supply of fatty acids required for mitochondrial ATP production. Inadequate LPL activity—seen in advanced diabetes—leads to energy deficiency, lipid accumulation, and ultimately, diabetic cardiomyopathy. Conversely, too much LPL activity in the vascular immune system, especially within macrophages, contributes to foam cell formation and plaque development.
This dual role demands a precision medicine approach—one that doesn’t attempt to universally boost or inhibit LPL, but instead modulates it based on tissue type and disease state. Advances in gene-editing, targeted drug delivery, and antisense oligonucleotide technologies could make this vision a reality. Imagine therapies that enhance cardiac LPL activity to support energy metabolism in the diabetic heart while silencing LPL expression in macrophages to prevent atherosclerosis. This tailored strategy could revolutionize the management of cardiometabolic diseases.
Moreover, the emerging success of GIP- and GLP-1–based therapies, as well as APOC3 and ANGPTL inhibitors, shows that lipid-centric approaches can be as impactful as glucose-lowering ones—especially when cardiovascular outcomes are the ultimate goal.
Looking forward, LPL is not just a metabolic enzyme—it is a therapeutic axis that integrates nutrition, energy use, and vascular health. As researchers continue to uncover its intricacies, LPL may hold the key to breaking the cycle of metabolic disease and cardiovascular failure, offering hope for millions at risk.
References
Shang, R., & Rodrigues, B. (2024). Lipoprotein lipase as a target for obesity/diabetes related cardiovascular disease. Journal of Pharmacy & Pharmaceutical Sciences, 27, 13199.
https://doi.org/10.3389/jpps.2024.13199
Lopaschuk, G. D., Ussher, J. R., Folmes, C. D., Jaswal, J. S., & Stanley, W. C. (2010). Myocardial fatty acid metabolism in health and disease. Physiological Reviews, 90(1), 207–258.
https://doi.org/10.1152/physrev.00015.2009
Goldberg, I. J., Eckel, R. H., & McPherson, R. (2011). Triglycerides and heart disease: Still a hypothesis? Arteriosclerosis, Thrombosis, and Vascular Biology, 31(8), 1716–1725.
https://doi.org/10.1161/ATVBAHA.111.226100
Puri, K., Lal, N., Shang, R., Ghosh, S., Flibotte, S., Dyer, R., et al. (2019). Diabetes mellitus severity and a switch from using lipoprotein lipase to adipose-derived fatty acid results in a cardiac metabolic signature that embraces cell death. Journal of the American Heart Association, 8(21), e014022.
https://doi.org/10.1161/JAHA.119.014022
Beigneux, A. P., Allan, C. M., Sandoval, N. P., Cho, G. W., Heizer, P. J., Jung, R. S., et al. (2019). Lipoprotein lipase is active as a monomer. Proceedings of the National Academy of Sciences, 116(13), 6319–6328.
https://doi.org/10.1073/pnas.1900983116
Young, S. G., Davies, B. S. J., Fong, L. G., & Beigneux, A. P. (2012). GPIHBP1: A platform for the lipolytic processing of triglyceride-rich lipoproteins. Current Opinion in Lipidology, 23(1), 35–43.
https://doi.org/10.1097/MOL.0b013e32834d2e49
Kim, M. S., Wang, F., Puthanveetil, P., Kewalramani, G., Hosseini-Beheshti, E., & Rodrigues, B. (2008). Protein kinase D is a key regulator of cardiomyocyte lipoprotein lipase secretion after diabetes. Circulation Research, 103(3), 252–260.
https://doi.org/10.1161/CIRCRESAHA.108.178681
Takahashi, M., Yagyu, H., Tazoe, F., Nagashima, S., Ohshiro, T., Okada, K., et al. (2013). Macrophage lipoprotein lipase modulates the development of atherosclerosis but not adiposity. Journal of Lipid Research, 54(4), 1124–1134.
https://doi.org/10.1194/jlr.M035568
Allahverdian, S., Chehroudi, A. C., McManus, B. M., Abraham, T., & Francis, G. A. (2014). Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation, 129(15), 1551–1559.
https://doi.org/10.1161/CIRCULATIONAHA.113.005015
Ichikawa, T., Liang, J., Kitajima, S., Koike, T., Wang, X., Sun, H., et al. (2005). Macrophage-derived lipoprotein lipase increases aortic atherosclerosis in cholesterol-fed Tg rabbits. Atherosclerosis, 179(1), 87–95.
https://doi.org/10.1016/j.atherosclerosis.2004.10.044
Kim, S. J., Nian, C., & McIntosh, C. H. (2010). GIP increases human adipocyte LPL expression through CREB and TORC2-mediated trans-activation of the LPL gene. Journal of Lipid Research, 51(11), 3145–3157.
https://doi.org/10.1194/jlr.M006841
Fong, L. G., Young, S. G., Beigneux, A. P., Bensadoun, A., & Oberer, M. (2016). GPIHBP1 and plasma triglyceride metabolism. Trends in Endocrinology & Metabolism, 27(7), 455–469.