GSK-3 and Neurodegeneration: A Molecular Target Redefining Alzheimer’s and Parkinson’s Therapies
Abstract
Glycogen Synthase Kinase-3 (GSK-3) has emerged as a critical enzyme in the pathogenesis of neurodegenerative diseases, particularly Alzheimer’s disease (AD) and Parkinson’s disease (PD). This blog explores the dual role of GSK-3 in promoting tau hyperphosphorylation and amyloid-β production in AD, as well as its involvement in α-synuclein toxicity in PD. It reviews the therapeutic potential of lithium, a well-known GSK-3 inhibitor, highlighting its preclinical benefits and mixed clinical outcomes. The article also outlines current challenges and future directions for developing safer, isoform-specific GSK-3-targeted therapies. As research advances, GSK-3 continues to represent a promising but complex target for disease-modifying interventions in neurodegenerative disorders.
Introduction — Why GSK-3 Matters in Neurodegeneration
Neurodegenerative diseases like Alzheimer’s disease (AD) and Parkinson’s disease (PD) represent some of the most challenging and costly public health crises of the 21st century. Despite decades of research, current treatments only offer modest symptomatic relief, with no disease-modifying therapies approved to halt or reverse the underlying progression. A growing body of evidence now points to a critical molecular player at the intersection of these disorders: Glycogen Synthase Kinase-3 (GSK-3).
GSK-3 is a serine/threonine protein kinase that plays a pivotal role in numerous physiological processes, including cell proliferation, neuroplasticity, and metabolism. It exists in two main isoforms—GSK-3α and GSK-3β—with GSK-3β being particularly prominent in the brain. Under normal conditions, GSK-3 is tightly regulated; however, in neurodegenerative diseases, this regulation appears to break down, leading to aberrant kinase activity.
What makes GSK-3 especially intriguing is its dual pathological relevance to both AD and PD. In Alzheimer’s disease, GSK-3β contributes to the hyperphosphorylation of tau protein, which promotes the formation of neurofibrillary tangles. Simultaneously, GSK-3 also plays a role in the production and toxicity of amyloid-β (Aβ) peptides—another key hallmark of AD. In Parkinson’s disease, GSK-3 is involved in the phosphorylation and aggregation of α-synuclein, the primary component of Lewy bodies, which are toxic to dopaminergic neurons.
Given this multifaceted involvement, GSK-3 has emerged as a compelling therapeutic target. Inhibiting its activity could offer a two-pronged approach to tackling the pathological drivers of multiple neurodegenerative diseases. Although many questions remain regarding the enzyme’s exact mechanisms and optimal modulation strategies, targeting GSK-3 offers a hopeful avenue for developing disease-modifying treatments in neurology.
GSK-3 and Alzheimer’s Disease — The Dual Role with Tau and Amyloid-β
Alzheimer’s disease (AD) is marked by two defining pathological features: intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein, and extracellular amyloid-β (Aβ) plaques derived from the amyloid precursor protein (APP). Emerging evidence highlights Glycogen Synthase Kinase-3 (GSK-3)—particularly the β isoform—as a central mediator of both these hallmark pathologies.
Tau is a microtubule-associated protein whose stability and function depend on precise phosphorylation. GSK-3β phosphorylates tau at multiple sites, and under pathological conditions, this activity becomes dysregulated. The result is tau hyperphosphorylation, which causes tau to detach from microtubules and aggregate into neurotoxic tangles. Notably, GSK-3β has been found both colocalized with hyperphosphorylated tau and capable of inducing tau pathology in transgenic animal models. Inhibiting GSK-3 activity reduces tau phosphorylation, restores microtubule function, and ameliorates neuronal deficits in several studies.
GSK-3 also plays a pivotal role in Aβ production and toxicity. It phosphorylates components of the γ-secretase complex and APP itself, promoting Aβ generation. Moreover, Aβ exposure can activate GSK-3β, which in turn exacerbates tau phosphorylation—a pathological feedback loop that accelerates neurodegeneration. Intriguingly, tau knockout models are protected from Aβ-induced toxicity, reinforcing the interdependence of these two pathways with GSK-3 at their core.
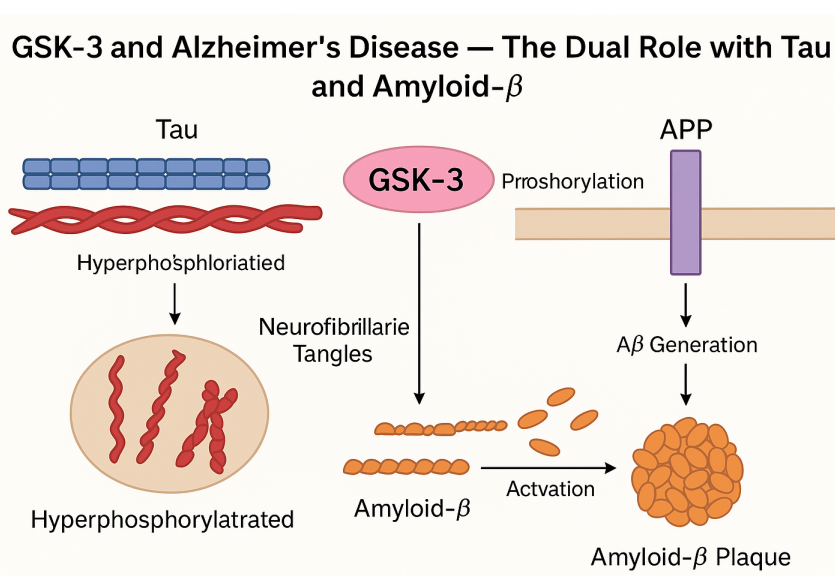
Fig.1 GSK-3 in Alzheimer’s: Tau and Amyloid-β Pathways
Given its involvement in both upstream (Aβ) and downstream (tau) pathways, GSK-3 serves as a molecular bridge between the two major mechanisms of AD progression. This makes it a highly attractive target for drug development, especially for therapies aiming to disrupt the interaction between Aβ accumulation and tau dysfunction. The dual role of GSK-3 in AD pathogenesis represents a promising frontier for developing more effective, mechanism-based interventions.
GSK-3’s Link to Parkinson’s Disease and α-Synuclein Toxicity
While Alzheimer’s disease (AD) research has long focused on GSK-3 due to its interaction with tau and amyloid-β, Parkinson’s disease (PD) has also emerged as a field where GSK-3 plays a significant role—particularly through its relationship with α-synuclein, a key pathological protein in PD. Parkinson’s disease is characterized by the progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta and the presence of Lewy bodies, which are rich in aggregated α-synuclein.
GSK-3β has been detected within Lewy bodies in PD brains and is upregulated in the striatum of PD patients. In addition to being a substrate for GSK-3β, α-synuclein may also regulate GSK-3β activity, creating a pathological feedback loop. Experimental studies show that in α-synuclein knockout models, GSK-3β activation is impaired, suggesting a reciprocal relationship between the two.
Importantly, GSK-3β contributes to the phosphorylation and aggregation of α-synuclein, a process believed to increase its neurotoxicity. This effect is supported by models using 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a neurotoxin that replicates Parkinsonian neurodegeneration. Inhibiting GSK-3β in MPTP-treated animals significantly reduces neuronal loss and decreases α-synuclein expression, indicating that GSK-3 activity directly influences disease severity.
These findings suggest that GSK-3β may act as a central regulator of multiple PD-related pathological processes, from α-synuclein phosphorylation to mitochondrial dysfunction and apoptosis. As such, GSK-3 inhibition represents a promising therapeutic strategy—not only for protecting vulnerable dopaminergic neurons but also for potentially halting the progression of α-synuclein pathology.
However, the clinical translation of these findings is still in early stages, and more studies are needed to determine whether GSK-3 inhibition can safely and effectively modify the course of PD in humans.
Lithium and GSK-3 Inhibition — Hope or Hype?
Among the various compounds investigated for modulating GSK-3 activity, lithium stands out as one of the earliest and most widely studied. Traditionally used as a mood stabilizer for bipolar disorder, lithium is now being explored for its neuroprotective properties, particularly through its inhibition of GSK-3. Given the central role of GSK-3β in Alzheimer’s and Parkinson’s disease pathology, lithium has been proposed as a candidate for repurposing in neurodegenerative therapeutics.
Lithium inhibits GSK-3 through two main mechanisms: direct competitive inhibition with magnesium ions (Mg²⁺) and indirect modulation via Akt and other upstream kinases. In cell and animal models, lithium has shown the ability to reduce tau phosphorylation, decrease amyloid-β (Aβ) production, and protect neurons from Aβ- and α-synuclein-induced toxicity. These effects make lithium a potential multitargeted agent, especially for disorders like Alzheimer’s disease, where both tau and Aβ are involved.
However, the clinical translation of these findings has produced mixed results. Some studies report cognitive benefits and reductions in biomarkers such as phosphorylated tau, while others find no significant improvement in cognitive function or disease progression. In a notable 10-week placebo-controlled trial, lithium treatment failed to produce statistically meaningful changes in CSF biomarkers or cognitive assessments, though an increase in brain-derived neurotrophic factor (BDNF) was observed in a subset of patients.
Furthermore, lithium’s narrow therapeutic window and potential side effects (including renal and thyroid dysfunction) complicate its long-term use, especially in elderly populations prone to comorbidities. In Parkinson’s disease models, lithium has shown neuroprotective effects in some cases but has also been associated with decreased dopamine release and ineffectiveness in halting dopaminergic neuron loss.
Thus, while lithium remains a valuable research tool and proof-of-concept agent, its clinical utility in neurodegenerative diseases is still under debate. The search continues for more selective, safer GSK-3 inhibitors that can reproduce lithium’s beneficial effects without its drawbacks.
Future Directions — Towards Better GSK-3-Targeted Therapies
The accumulating evidence that Glycogen Synthase Kinase-3 (GSK-3) plays a central role in neurodegenerative diseases has catalyzed significant interest in developing targeted GSK-3 inhibitors. However, despite promising results in preclinical models, the translation of GSK-3 inhibition into successful clinical therapies remains elusive. This disconnect emphasizes the need to refine our approach and develop next-generation therapeutics with improved specificity, safety, and mechanistic insight.
One major challenge is the complex regulatory network surrounding GSK-3, which is involved in numerous physiological pathways beyond the brain, including insulin signaling, inflammation, and cell survival. Systemic inhibition of GSK-3—particularly GSK-3β—risks widespread off-target effects. Thus, future efforts should focus on isoform-specific inhibitors that can differentiate between GSK-3α and GSK-3β, and ideally modulate their activity in a tissue-selective or pathology-dependent manner.
Another priority is understanding the contextual role of GSK-3 in disease progression. In Alzheimer’s disease, GSK-3 activity appears to promote both amyloid and tau pathology. In Parkinson’s disease, it modulates α-synuclein phosphorylation and toxicity. Yet, GSK-3 may also have protective functions under certain conditions, suggesting that complete inhibition could be counterproductive. This underscores the need for modulators rather than blunt inhibitors—agents that can restore balance to GSK-3 signaling without total shutdown.
In addition, biomarker development will be essential to guide GSK-3-targeted therapies. Monitoring kinase activity in accessible tissues like blood or CSF, along with tau and amyloid biomarkers, could help identify responsive patients and optimize dosing strategies.
Emerging approaches such as allosteric modulators, peptide-based inhibitors, or RNA-targeted therapies are also under investigation. As our understanding of GSK-3’s structural biology and interactome grows, so too does the potential to design highly selective drugs that intervene at the right place, time, and dose.
Ultimately, the therapeutic future of GSK-3 will depend not just on inhibiting an enzyme, but on fine-tuning a complex signaling hub central to brain health and disease.
References
Lei, P., Ayton, S., Bush, A. I., & Adlard, P. A. (2011). GSK‐3 in neurodegenerative diseases. International Journal of Alzheimer’s Disease, 2011, 1–9.
https://doi.org/10.4061/2011/189246
Frame, S., & Cohen, P. (2001). GSK3 takes centre stage more than 20 years after its discovery. Biochemical Journal, 359(1), 1–16.
https://doi.org/10.1042/bj3590001
Phiel, C. J., Wilson, C. A., Lee, V. M. Y., & Klein, P. S. (2003). GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature, 423(6938), 435–439.
https://doi.org/10.1038/nature01640
Duka, T., Duka, V., Joyce, J. N., & Sidhu, A. (2009). α-Synuclein contributes to GSK-3β-catalyzed tau phosphorylation in Parkinson’s disease models. FASEB Journal, 23(9), 2820–2830.
https://doi.org/10.1096/fj.08-126979
Hampel, H., Ewers, M., Burger, K., et al. (2009). Lithium trial in Alzheimer’s disease: A randomized, single-blind, placebo-controlled, multicenter 10-week study. Journal of Clinical Psychiatry, 70(6), 922–931.
https://doi.org/10.4088/JCP.08m04594
Sereno, L., Coma, M., Rodríguez, M., et al. (2009). A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiology of Disease, 35(3), 359–367.