Growth Factor Signaling and the Cell Cycle: How ERK and AKT Pathways Drive Proliferation
Abstract
Cell division is a tightly regulated process that integrates external cues with internal molecular machinery. Among the most important regulators are growth factors (GFs), which signal through receptor tyrosine kinases (RTKs) to activate pathways such as Ras/ERK and PI3K/AKT. These pathways govern the activity of cyclins, cyclin-dependent kinases (CDKs), and inhibitors that determine whether a cell progresses through the G1 restriction point and commits to DNA replication. Recent findings highlight the importance of timing and dynamics, with evidence that two distinct signaling pulses in G1 encode the decision to divide. Moreover, emerging studies suggest that growth factor signaling may extend its influence beyond G1, affecting S phase, G2, and mitosis, although these roles remain less clearly defined. Understanding how extracellular signals orchestrate cell-cycle control is vital not only for basic biology but also for clinical applications, particularly in cancer, where deregulated ERK and AKT activity drive unchecked proliferation.
Introduction: Growth Factors as the Cell’s Decision-Makers
Cells are the fundamental units of life, and one of their most critical functions is the ability to divide. However, cell division is not a process that occurs spontaneously or without regulation. For a cell to progress through its cycle and produce daughter cells, it must first receive the right signals from its environment. Among the most influential of these signals are growth factors (GFs), a group of extracellular proteins that act like “decision-makers,” instructing cells when and how to proliferate.
Growth factors exert their influence primarily through receptor tyrosine kinases (RTKs) located on the cell surface. When a growth factor binds to its receptor, it triggers receptor dimerization and autophosphorylation, setting off a cascade of intracellular signaling events. These events converge on pathways such as Ras/ERK and PI3K/AKT, which ultimately influence whether a cell enters the cell cycle, remains in a quiescent state, or undergoes differentiation. This intricate system ensures that cells divide only when conditions are favorable, preventing uncontrolled growth that could lead to diseases like cancer.
The importance of growth factors can be appreciated by considering the first stage of the cell cycle, known as G1 phase. At this stage, the cell is essentially waiting for a “go-ahead” signal. If growth factors are absent, cells may enter a resting state (G0) where they remain metabolically active but non-dividing. However, when growth factors are present, they activate the necessary signaling pathways to induce the expression of cyclins and cyclin-dependent kinases (CDKs), the molecular engines that drive cell-cycle progression. This is a crucial checkpoint: without growth factor input, the cell will not commit to DNA replication and division.
From an evolutionary perspective, this dependence on extracellular signals provides a major survival advantage. By linking cell division to environmental cues such as nutrient availability, tissue growth demands, and repair signals, organisms maintain tissue homeostasis and avoid unnecessary energy expenditure. Moreover, growth factor regulation ensures that proliferation is tightly coordinated with the needs of the organism, rather than occurring randomly.
Understanding how growth factors orchestrate these processes has profound implications for medicine. Dysregulation of growth factor signaling is a hallmark of many cancers, where cells escape normal control and proliferate unchecked. At the same time, harnessing growth factors has therapeutic potential in regenerative medicine and wound healing. Thus, studying these molecular decision-makers is central to both fundamental biology and clinical innovation.
The Cell Cycle’s Gatekeepers: Cyclins, CDKs, and Checkpoints
The cell cycle is the highly coordinated sequence of events that allows a single cell to grow, replicate its DNA, and divide into two daughter cells. To maintain order, cells rely on molecular “gatekeepers” that regulate when each phase begins and ends. At the core of this control system are cyclins and cyclin-dependent kinases (CDKs), which act as the engines driving the cycle forward. Equally important are cell-cycle checkpoints, surveillance mechanisms that ensure division occurs only under proper conditions. Together, these regulators guarantee both accuracy and fidelity in cell proliferation.
Cyclins are regulatory proteins whose concentrations rise and fall at specific stages of the cell cycle. They bind to CDKs, activating them to phosphorylate downstream targets that enable transitions between phases. For example, Cyclin D/CDK4-6 complexes drive progression through early G1, while Cyclin E/CDK2 promotes passage through the restriction point and commitment to DNA synthesis. During S phase, Cyclin A/CDK2 supports DNA replication, and Cyclin B/CDK1 is essential for entry into mitosis. This sequential activation ensures that events occur in a precise order, avoiding conflicts such as attempting DNA replication before repair is complete.
Checkpoints serve as the brakes of the cycle, halting progression when errors or unfavorable conditions are detected. The G1/S checkpoint ensures that DNA damage is repaired before replication begins. The G2/M checkpoint prevents entry into mitosis if DNA has not been fully replicated or contains damage. Finally, the spindle assembly checkpoint (SAC) during mitosis ensures that chromosomes are correctly attached to spindle fibers before segregation occurs. These safety nets are crucial, as unchecked progression can result in genomic instability—a hallmark of cancer.
Growth factor (GF) signaling integrates with cyclin–CDK activity at these checkpoints. Without mitogenic cues from the environment, cells will not accumulate sufficient cyclin levels to cross the restriction point in late G1. In this way, external signals are translated into internal machinery, tightly coupling cell proliferation to tissue and organismal needs.
Understanding cyclin–CDK regulation and checkpoint control has major clinical implications. Many cancers harbor mutations in CDKs, cyclins, or checkpoint regulators such as p53 and Rb, leading to loss of control over proliferation. This insight has fueled the development of CDK inhibitors, a growing class of anticancer drugs that target these core engines of the cell cycle.
Signal Integration in G1: Ras/ERK and PI3K/AKT at Work
The G1 phase of the cell cycle is the most critical decision-making point, where cells determine whether to commit to division, pause, or exit into a quiescent state. At this stage, external growth factors (GFs) transmit signals through receptor tyrosine kinases (RTKs), activating intracellular cascades that converge on two master pathways: Ras/ERK and PI3K/AKT. These pathways coordinate the expression and stability of cyclins and cyclin-dependent kinase (CDK) regulators, effectively translating extracellular cues into cell-cycle progression.
The Ras/ERK pathway plays a central role in promoting G1 progression. When activated, ERK translocates into the nucleus and induces the expression of Cyclin D1, a key activator of CDK4/6. ERK also stabilizes the transcription factor c-Myc, which enhances the transcription of numerous cell-cycle genes. In addition, ERK signaling transiently induces p21^Cip1, a dual-function protein that can both facilitate Cyclin D/CDK4-6 activity and, later, inhibit CDK2 activity to maintain balance. ERK also contributes to the downregulation of p27^Kip1, a CDK inhibitor whose removal from the nucleus is required for cells to move past the G1 restriction point. Together, these effects ensure that the molecular machinery necessary for S phase entry is primed.
In parallel, the PI3K/AKT pathway provides complementary and overlapping signals. AKT activation stabilizes p21^Cip1, but it also phosphorylates p27^Kip1 at threonine 157, driving it out of the nucleus and thereby reducing its inhibitory effect on CDKs. AKT additionally inactivates GSK3, which prevents the degradation of Cyclin D1, thereby sustaining CDK4/6 activity. Moreover, AKT signaling can promote the induction of c-Fos, another transcriptional regulator linked to proliferation. Importantly, the tumor suppressor PTEN antagonizes PI3K/AKT activity by dephosphorylating PIP3, highlighting how balance between activation and inhibition is essential to avoid uncontrolled growth.
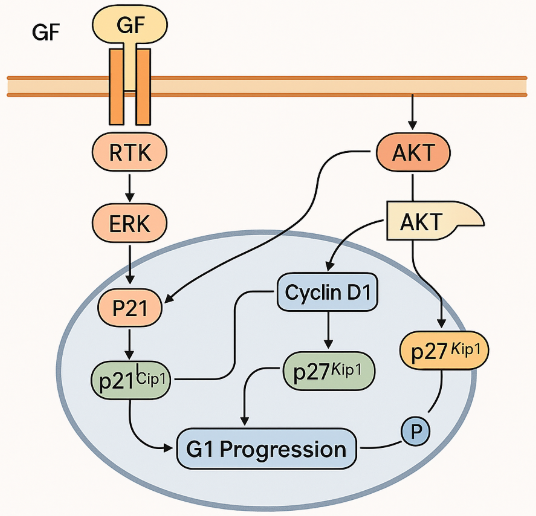
Figure 1. ERK and AKT: Teamwork in G1 Cell Cycle Control
What makes the G1 phase especially interesting is not just the independent roles of ERK and AKT, but their crosstalk and convergence. Both pathways regulate overlapping targets—p21, p27, and Cyclin D1—ensuring robust commitment to DNA replication only when external conditions are favorable. This redundancy also provides resilience: if one pathway is partially inhibited, the other can compensate. Such integration is vital in normal physiology, but it also explains why cancers frequently exhibit hyperactivation of both Ras/ERK and PI3K/AKT, allowing them to bypass growth control.
By orchestrating the balance of cyclins and CDK inhibitors, Ras/ERK and PI3K/AKT signaling in G1 transform external growth factor input into an irreversible commitment to cell division.
Timing is Everything: The Two-Pulse Model of G1 Signaling
Cell proliferation is not merely a matter of having growth factors (GFs) present—it is also about the timing and dynamics of the signals they generate. Recent research has revealed that, even under continuous GF exposure, cells in the G1 phase do not respond with a steady signal. Instead, they display a two-pulse model of Ras/ERK and PI3K/AKT activity. These distinct bursts of signaling act as a molecular code, guiding the cell’s decision to commit to DNA replication.
The first pulse occurs as cells transition from quiescence (G0) into early G1. This early transient activation of Ras/ERK and PI3K/AKT primes the cell for growth by initiating expression of Cyclin D1 and preparing CDK4/6 for activity. It essentially provides the initial “wake-up call,” ensuring that the cell exits its resting state and starts moving toward the restriction point. Without this initial surge, cells are unlikely to leave G0, even if growth factors remain present.
The second pulse happens later in G1, closer to the restriction point. This sustained activation reinforces the cell’s commitment by stabilizing Cyclin D1, downregulating CDK inhibitors like p27, and ensuring CDK2 activity for S-phase entry. This later wave is essential for passing the critical checkpoint where the cell decides whether to replicate its DNA. If the second pulse is absent or weak, cells may stall in G1 or re-enter quiescence instead of dividing.
An intriguing discovery is that two short bursts of growth factor exposure can substitute for continuous stimulation, provided they are spaced correctly to mimic the natural two-pulse pattern. This suggests that cells interpret mitogenic signals in a dynamic, time-dependent manner rather than simply responding to their concentration. Such dynamics add an extra layer of regulation, ensuring that proliferation is not triggered by random or transient stimuli but by sustained and well-coordinated signaling events.
The two-pulse model has significant implications for cancer biology. Many tumors display deregulated ERK or AKT activity, which can override the natural timing requirements and push cells through G1 inappropriately. Understanding how pulse dynamics work may help explain why some cancer therapies that block growth factor receptors or downstream kinases succeed, while others fail. It also opens opportunities for novel therapeutic approaches that disrupt the timing of signals rather than just their intensity.
Beyond G1: Emerging Roles and Open Questions
While the G1 phase is the most thoroughly understood point of growth factor (GF) control, evidence increasingly suggests that Ras/ERK and PI3K/AKT signaling may also influence later phases of the cell cycle. These roles are less clearly defined than in G1, but they point to additional layers of complexity in how external cues integrate with intracellular machinery.
In S phase, where DNA replication occurs, GF signaling may help regulate replication timing and maintain genomic stability. Some studies have shown that ERK activation can influence replication origin firing, while PI3K/AKT signaling supports nucleotide synthesis and DNA repair. Disruption of these processes can lead to replication stress, a feature commonly observed in cancer cells.
During G2 phase, the period between DNA replication and mitosis, Ras/ERK and PI3K/AKT activity have been implicated in the regulation of Cyclin B/CDK1, the key driver of mitotic entry. For instance, ERK signaling has been reported to fine-tune the G2/M transition by modulating Cdc25 phosphatases, which activate CDK1. Similarly, AKT can influence G2 progression by promoting recovery from DNA damage checkpoints, thereby allowing cells to enter mitosis once repairs are complete. However, these effects appear to vary depending on cell type and context, highlighting the need for further investigation.
In M phase (mitosis), GF signaling seems to undergo significant rewiring. EGFR-dependent signaling, for example, can become uncoupled from ERK but maintain AKT activity, particularly via Akt2. This may ensure survival signals during the stressful process of chromosome segregation. The precise role of these pathways in spindle checkpoint control and chromosome segregation remains a matter of debate, but their involvement suggests that GF signaling extends well beyond the G1 restriction point.
These emerging findings raise important open questions. Do growth factor signals play an active role throughout all phases of the cycle, or are their effects primarily indirect, mediated by changes established in G1? Are the later-phase functions of ERK and AKT universal features of cell-cycle control, or are they context-dependent, relevant mainly in stressed or transformed cells? Answering these questions is not only key for basic biology but also crucial for cancer therapy. Many targeted drugs inhibit ERK or AKT signaling, but their success may hinge on understanding how timing and phase-specific regulation influence treatment outcomes.
References
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646–674.
https://doi.org/10.1016/j.cell.2011.02.013
Lemmon, M. A., & Schlessinger, J. (2010). Cell signaling by receptor tyrosine kinases. Cell, 141(7), 1117–1134.
https://doi.org/10.1016/j.cell.2010.06.011
Massagué, J. (2004). G1 cell-cycle control and cancer. Nature, 432(7015), 298–306.
https://doi.org/10.1038/nature03094
Yarden, Y., & Pines, G. (2012). The ERBB network: At last, cancer therapy meets systems biology. Nature Reviews Cancer, 12(8), 553–563.
https://doi.org/10.1038/nrc3309
Sherr, C. J., & Roberts, J. M. (1999). CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes & Development, 13(12), 1501–1512.
https://doi.org/10.1101/gad.13.12.1501
Nurse, P. (2000). A long twentieth century of the cell cycle and beyond. Cell, 100(1), 71–78.
https://doi.org/10.1016/S0092-8674(00)81687-6
Malumbres, M., & Barbacid, M. (2009). Cell cycle, CDKs and cancer: A changing paradigm. Nature Reviews Cancer, 9(3), 153–166.
https://doi.org/10.1038/nrc2602
Dhillon, A. S., Hagan, S., Rath, O., & Kolch, W. (2007). MAP kinase signalling pathways in cancer. Oncogene, 26(22), 3279–3290.
https://doi.org/10.1038/sj.onc.1210421
Porta, C., Paglino, C., & Mosca, A. (2014). Targeting PI3K/Akt/mTOR signaling in cancer. Frontiers in Oncology, 4, 64.
https://doi.org/10.3389/fonc.2014.00064
Spencer, S. L., Cappell, S. D., Tsai, F. C., Overton, K. W., Wang, C. L., & Meyer, T. (2013). The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell, 155(2), 369–383.
https://doi.org/10.1016/j.cell.2013.08.062
Purvis, J. E., & Lahav, G. (2013). Encoding and decoding cellular information through signaling dynamics. Cell, 152(5), 945–956.
https://doi.org/10.1016/j.cell.2013.02.005
Blagosklonny, M. V., & Pardee, A. B. (2002). The restriction point of the cell cycle. Cell Cycle, 1(2), 103–110.
https://doi.org/10.4161/cc.1.2.108
Doménech, E., & Malumbres, M. (2013). Mitosis-targeted anti-cancer therapies: We still need to know more. Cell Cycle, 12(9), 1501–1502.
https://doi.org/10.4161/cc.24932
Liu, P., Begley, M., Michowski, W., Inuzuka, H., Ginzberg, M., Gao, D., … & Cantley, L. C. (2014). Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature, 508(7497), 541–545.
https://doi.org/10.1038/nature13079
Krenning, L., Feringa, F. M., Shaltiel, I. A., van den Berg, J., & Medema, R. H. (2014). Transient activation of p53 in G2 phase is sufficient to induce senescence. Molecular Cell, 55(1), 59–72.