Epigenetics and Cancer Stem Cells: Unlocking the Future of Precision Oncology
Abstract
Cancer stem cells (CSCs) are a rare but critical subpopulation of tumor cells that drive tumor initiation, metastasis, therapy resistance, and relapse. Recent research highlights the pivotal role of epigenetic mechanisms—including DNA methylation, histone modification, and chromatin remodeling—in sustaining CSC identity and function. These modifications not only regulate oncogenic pathways like Wnt, Hedgehog, and Notch but also shield CSCs from immune detection and cytotoxic treatments. This blog explores the current understanding of epigenetic regulation in CSCs and examines how emerging epigenetic therapies—from DNMT and HDAC inhibitors to EZH2 and BET bromodomain inhibitors—are reshaping the landscape of precision oncology. By integrating epigenetics with immunotherapy and molecular profiling, a new era of targeted cancer treatment is on the horizon.
Cracking the Code of Cancer Stem Cells
Despite decades of progress in cancer research, one of the most perplexing challenges remains: why do tumors often recur after initially successful treatment? The answer may lie in a rare but potent subpopulation of cells known as cancer stem cells (CSCs).
CSCs are not typical cancer cells. Much like normal stem cells, they possess the ability to self-renew and differentiate into various cell types. But their defining trait—and the one that makes them dangerous—is their tumor-initiating capacity. A small number of CSCs can regenerate an entire tumor, and even worse, survive therapies that destroy the bulk of cancer cells. These properties position CSCs as key drivers of tumor initiation, metastasis, resistance to chemotherapy, and ultimately, cancer relapse.
But what gives CSCs this supercharged resilience? While genetic mutations play a role, mounting evidence points to epigenetic regulation as the true puppet master. Unlike genetic mutations, which alter the DNA sequence itself, epigenetic changes modify how genes are expressed without changing the underlying code. Mechanisms such as DNA methylation, histone modification, and chromatin remodeling work in tandem to silence tumor suppressors, activate oncogenic pathways, and lock CSCs into a self-renewing, therapy-resistant state.
Understanding these epigenetic mechanisms is not just a matter of academic interest—it has transformative implications for treatment. By targeting the specific epigenetic alterations that sustain CSCs, researchers are uncovering new pathways to suppress tumor growth at its root, reduce recurrence, and enhance therapeutic sensitivity. In this blog series, we explore how these molecular-level insights are reshaping our approach to cancer, starting with the fundamental role of epigenetics in regulating cancer stem cells.
Epigenetic Mechanisms Behind CSC Behavior
Cancer stem cells (CSCs) owe much of their power to an invisible hand: epigenetic regulation. Unlike genetic mutations that irreversibly alter DNA sequences, epigenetic mechanisms act as molecular switches, dynamically turning genes on or off. This fine-tuned control is what allows CSCs to maintain their identity, survive hostile environments, and evade even the most aggressive therapies.
One of the most studied epigenetic mechanisms is DNA methylation—the addition of methyl groups to cytosine bases, typically at CpG islands near gene promoters. In CSCs, hypermethylation of tumor suppressor genes like p16, PTEN, and RB leads to their silencing, allowing cancer cells to proliferate unchecked. Conversely, hypomethylation of oncogenes can result in aberrant activation of pathways that promote self-renewal and metastasis.
Another crucial layer is histone modification. Histones are proteins around which DNA is wound, and their chemical tags—such as methylation, acetylation, and phosphorylation—alter chromatin structure and gene accessibility. For example, trimethylation at histone H3 lysine 4 (H3K4me3) is associated with active gene expression, whereas H3K27me3 correlates with repression. In CSCs, the balance of these modifications is often skewed to maintain genes that promote stemness and pluripotency, such as OCT4, SOX2, and NANOG.
Beyond methylation, chromatin remodeling complexes like SWI/SNF and Polycomb group proteins influence the spatial organization of DNA, determining whether genes remain silent or become activated. Abnormal function of these complexes in CSCs helps maintain a stem-like, undifferentiated state, often at the expense of normal tissue architecture and function.
Finally, non-coding RNAs, including microRNAs and long non-coding RNAs, add another epigenetic layer by post-transcriptionally regulating genes involved in cell cycle control, apoptosis, and differentiation. Their dysregulation is frequently observed in CSC populations.
Together, these mechanisms build a highly adaptive epigenetic landscape—one that sustains CSC behavior and creates new opportunities for targeted therapies.
Epigenetic Hijacking of Core Signaling Pathways
Cancer stem cells (CSCs) don’t operate in isolation—they are tightly intertwined with major oncogenic signaling pathways that regulate self-renewal, differentiation, and survival. What makes CSCs particularly formidable is how epigenetic modifications hijack these pathways, locking them into a hyperactive state that fuels tumor progression and therapeutic resistance.
One of the most prominent pathways influenced by epigenetics is the Wnt/β-catenin pathway, essential for embryonic development and stem cell maintenance. In CSCs, this pathway is frequently overactivated due to epigenetic silencing of Wnt antagonists such as DKK1 and SFRP1. Aberrant methylation of their promoter regions allows unchecked Wnt signaling, sustaining the expression of stemness genes like c-MYC and Cyclin D1.
The Hedgehog (Hh) pathway, another developmental signaling cascade, is similarly co-opted. Normally quiescent in adult tissues, Hedgehog signaling becomes abnormally activated in CSCs through hypomethylation of the Shh promoter, or through altered chromatin states mediated by histone deacetylases (HDACs). This activation enhances proliferation and self-renewal, particularly in brain, colon, and pancreatic cancers.
The Notch pathway, responsible for cell fate determination, is also frequently dysregulated in CSCs. Histone methylation and acetylation of Notch target genes, such as HES1 and HEY1, result in sustained pathway activity. Furthermore, the Polycomb group protein EZH2, a histone methyltransferase, works in tandem with Notch to repress differentiation genes, further entrenching the stem-like state.
These epigenetically modified pathways do not act independently—they form a dynamic regulatory network. Cross-talk among Wnt, Hedgehog, and Notch amplifies CSC traits and supports immune evasion, angiogenesis, and resistance to apoptosis. This multilayered control not only preserves the CSC population but also shapes the broader tumor ecosystem.
Targeting these epigenetically dysregulated pathways offers a dual advantage: disrupting CSC maintenance and weakening the tumor’s regenerative capacity.
Targeting CSCs with Epigenetic Therapy
The elusive nature of cancer stem cells (CSCs) presents a major hurdle in cancer therapy. Their resilience to chemotherapy and radiotherapy stems not only from their intrinsic stem-like properties but also from epigenetic reprogramming that shields them from cellular death and promotes long-term survival. Recognizing this, researchers have turned to epigenetic therapies—drugs designed to reverse abnormal gene expression patterns and re-sensitize CSCs to conventional treatments.
Among the most advanced classes of epigenetic drugs are DNA methyltransferase inhibitors (DNMTis) such as azacitidine and decitabine. These agents demethylate tumor suppressor gene promoters, restoring expression of genes that inhibit self-renewal and promote differentiation. Initially approved for hematologic malignancies like myelodysplastic syndromes (MDS), these compounds are now under investigation for their capacity to deplete CSC populations across a range of solid tumors.
Equally promising are histone deacetylase inhibitors (HDACis) such as vorinostat and romidepsin. These drugs increase histone acetylation, leading to a more open chromatin structure and reactivation of silenced genes. In CSCs, HDACis have been shown to downregulate stemness markers (e.g., OCT4, SOX2), induce apoptosis, and sensitize cells to chemotherapy. Some studies also suggest that HDACis may disrupt the tumor microenvironment, making it less hospitable to CSCs.
In addition to these approved therapies, a new wave of targeted epigenetic inhibitors is entering clinical trials. These include EZH2 inhibitors (e.g., EPZ-6438) that block histone methyltransferase activity, LSD1 inhibitors targeting histone demethylation, and BET bromodomain inhibitors like JQ1 that suppress oncogenic transcription factors such as MYC. Early findings indicate that these drugs can impair CSC self-renewal, reduce tumorigenicity, and enhance immune recognition.
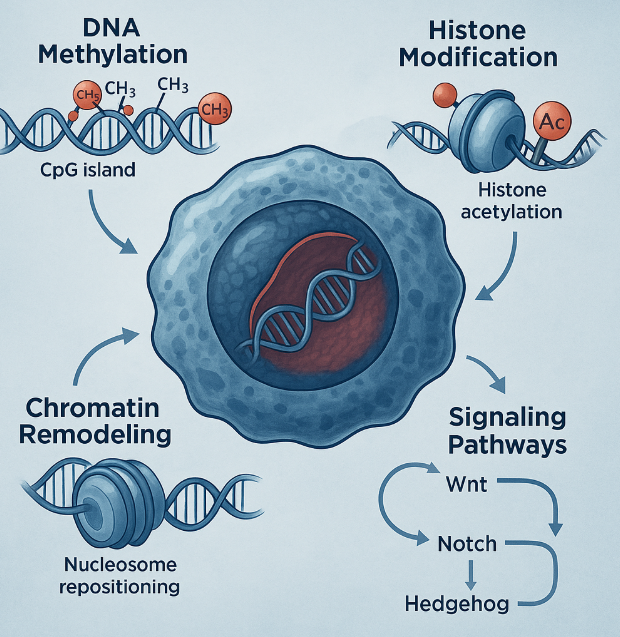
Fig. 1 Epigenetic Mechanisms Driving Cancer Stem Cell Regulation
Ultimately, the strength of epigenetic therapy lies in its potential to reprogram CSCs from a treatment-resistant state to one that is vulnerable—and curable. When combined with traditional therapies or immunotherapy, epigenetic drugs may finally tip the scales in favor of long-term remission.
Future Perspectives — A New Era in Precision Oncology
As the curtain lifts on a new era of cancer research, the integration of epigenetics into precision oncology stands at the forefront of innovation. With traditional therapies often failing to eliminate the most resilient cancer stem cells (CSCs), researchers are now turning to the epigenome—the regulatory blueprint of gene expression—as a therapeutic focal point. This shift is redefining how we approach cancer treatment, from one-size-fits-all protocols to targeted, adaptable strategies.
One of the most promising frontiers is the synergy between epigenetic therapy and immunotherapy. Emerging evidence suggests that epigenetic drugs can enhance immune responses by increasing the visibility of cancer cells. For instance, demethylating agents can upregulate tumor-associated antigens and MHC class I molecules, making CSCs more susceptible to immune recognition. Moreover, histone modifiers can influence PD-L1 expression, potentially improving the effectiveness of immune checkpoint inhibitors like anti-PD-1 and anti-CTLA-4 antibodies.
Another critical area of development is the personalization of epigenetic treatments. Advances in next-generation sequencing now allow clinicians to profile an individual tumor’s epigenetic landscape in remarkable detail. This opens the door to precision-guided therapies, where specific epigenetic abnormalities in CSCs can be matched with targeted inhibitors—offering a level of specificity previously unattainable.
Yet, challenges remain. Epigenetic changes are dynamic and context-dependent, and their effects can vary widely across tumor types and patient populations. Moreover, off-target effects and the reversibility of some epigenetic modifications necessitate cautious and strategic application. Ongoing clinical trials will be pivotal in validating the long-term efficacy and safety of these therapies.
Still, the momentum is undeniable. As we continue to unravel the epigenetic codes governing CSCs, the potential to disrupt cancer at its core becomes more tangible. By combining epigenetic insights with cutting-edge diagnostics and immunologic tools, we edge closer to a future where relapse is rare—and remission is permanent.
References
Reya, T., Morrison, S. J., Clarke, M. F., & Weissman, I. L. (2001). Stem cells, cancer, and cancer stem cells. Nature, 414(6859), 105–111. https://doi.org/10.1038/35102167
Toh, T. B., Lim, J. J., & Chow, E. K. H. (2017). Epigenetics in cancer stem cells. Molecular Cancer, 16(1), 29. https://doi.org/10.1186/s12943-017-0596-9
Jones, P. A., & Baylin, S. B. (2007). The epigenomics of cancer. Cell, 128(4), 683–692. https://doi.org/10.1016/j.cell.2007.01.029
Easwaran, H., Tsai, H. C., & Baylin, S. B. (2014). Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Molecular Cell, 54(5), 716–727. https://doi.org/10.1016/j.molcel.2014.05.015
Baylin, S. B., & Jones, P. A. (2011). A decade of exploring the cancer epigenome—Biological and translational implications. Nature Reviews Cancer, 11(10), 726–734. https://doi.org/10.1038/nrc3130
Widschwendter, M., & Jones, P. A. (2002). DNA methylation and breast carcinogenesis. Oncogene, 21(35), 5462–5482. https://doi.org/10.1038/sj.onc.1205606
Takebe, N., Miele, L., Harris, P. J., Jeong, W., Bando, H., Kahn, M., Yang, S. X., & Ivy, S. P. (2015). Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nature Reviews Clinical Oncology, 12(8), 445–464. https://doi.org/10.1038/nrclinonc.2015.61
Esteller, M. (2008). Epigenetics in cancer. New England Journal of Medicine, 358(11), 1148–1159. https://doi.org/10.1056/NEJMra072067
Katoh, M., & Katoh, M. (2007). WNT signaling pathway and stem cell signaling network. Clinical Cancer Research, 13(14), 4042–4045. https://doi.org/10.1158/1078-0432.CCR-07-0329
Griffiths, E. A., & Gore, S. D. (2008). Epigenetic therapies in MDS and AML. Advances in Experimental Medicine and Biology, 624, 39–60. https://doi.org/10.1007/978-0-387-77576-7_4
Eckschlager, T., Plch, J., Stiborova, M., & Hrabeta, J. (2017). Histone deacetylase inhibitors as anticancer drugs. International Journal of Molecular Sciences, 18(7), 1414. https://doi.org/10.3390/ijms18071414
Topper, M. J., Vaz, M., Chiappinelli, K. B., DeStefano Shields, C. E., Niknafs, N., Yen, R. C., … & Baylin, S. B. (2017). Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell, 171(6), 1284–1300.e21. https://doi.org/10.1016/j.cell.2017.10.022