Epigenetic Therapies in Cancer: Targeting Histone Methylation from Lab to Clinic
Abstract
Histone methylation is a pivotal epigenetic mechanism that regulates gene expression and chromatin structure. In cancer, dysregulation of histone methyltransferases and demethylases contributes to aberrant gene silencing, oncogene activation, and therapy resistance. This blog post explores the emerging landscape of epigenetic cancer therapy, focusing on key enzymatic targets—such as DOT1L, EZH2, and LSD1—and the small-molecule inhibitors currently undergoing clinical trials. It also highlights the therapeutic challenges related to selectivity, drug delivery, and resistance, while emphasizing future strategies including combination therapies, reader domain targeting, and personalized epigenomic profiling. As research advances, targeting histone methylation offers a promising avenue for more precise and reversible cancer treatment.
Introduction: Epigenetics Enters the Cancer Arena
Cancer has long been understood as a disease of genetic mutations—irreversible errors in DNA that drive uncontrolled cell growth. But a quieter revolution is underway in oncology, one that looks beyond the genetic code to the epigenetic landscape that governs gene expression. Epigenetics refers to heritable changes in gene function that occur without altering the underlying DNA sequence. These modifications can determine whether a gene is turned on or off, and they play a pivotal role in cellular identity, development, and disease.
One of the most influential epigenetic mechanisms involves chemical modifications to histone proteins—the molecular spools around which DNA is wound. These histone modifications, especially methylation, can either activate or repress gene transcription, depending on the site and context. Crucially, unlike genetic mutations, histone methylation is reversible, making it an attractive target for therapeutic intervention.
In cancer, epigenetic dysregulation is now recognized as a key contributor to tumor development, progression, and therapy resistance. Aberrant patterns of histone methylation can silence tumor suppressor genes or activate oncogenes, even in the absence of DNA mutations. Enzymes that add (methyltransferases) or remove (demethylases) methyl groups from histones are often found mutated, overexpressed, or misregulated in cancer cells.
This emerging understanding has ushered in a new wave of epigenetic drugs aimed at restoring balance to the cancer epigenome. Unlike traditional chemotherapy, which targets rapidly dividing cells indiscriminately, these drugs seek to precisely reprogram gene expression. By inhibiting specific histone-modifying enzymes, scientists hope to unlock a new class of targeted, reversible, and personalized therapies.
As of today, several such compounds are making their way through clinical trials, targeting key enzymes like DOT1L, EZH2, and LSD1. The era of epigenetic cancer therapy has arrived—and it’s just getting started.
The Role of Histone Methyltransferases and Demethylases
To understand how epigenetic therapies work, we must first meet their primary targets: the enzymes responsible for adding or removing methyl groups from histones. These enzymes are the writers and erasers of the histone code, shaping the chromatin landscape that controls gene expression.
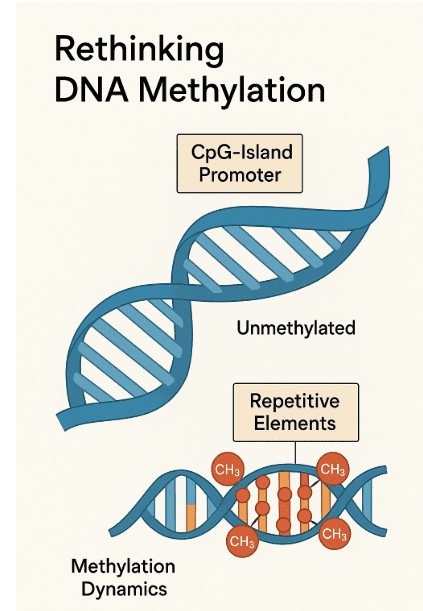
Histone methylation occurs mainly on lysine and arginine residues, with the methyltransferases (writers) adding methyl groups and demethylases (erasers) removing them. Depending on the location and degree (mono-, di-, or trimethylation), these modifications can either activate or repress gene transcription.
Among the best-studied methyltransferases is EZH2, the catalytic subunit of the polycomb repressive complex 2 (PRC2). EZH2 tri-methylates histone H3 at lysine 27 (H3K27me3), a repressive mark linked to gene silencing. In cancer, EZH2 is often overexpressed or mutated, leading to the silencing of tumor suppressor genes—particularly in lymphomas and solid tumors.
On the other end of the spectrum is DOT1L, a unique methyltransferase that methylates H3K79—a mark typically associated with gene activation. While DOT1L itself isn’t frequently mutated, its activity is hijacked in MLL-rearranged leukemias, where fusion proteins recruit DOT1L to aberrant gene loci, driving malignant transformation.
The demethylase LSD1 (KDM1A) serves as a potent epigenetic eraser. It primarily removes mono- and dimethyl groups from H3K4, a mark of active promoters. LSD1 overexpression has been linked to several cancers, including prostate cancer and acute myeloid leukemia, where it sustains undifferentiated, proliferative cell states.
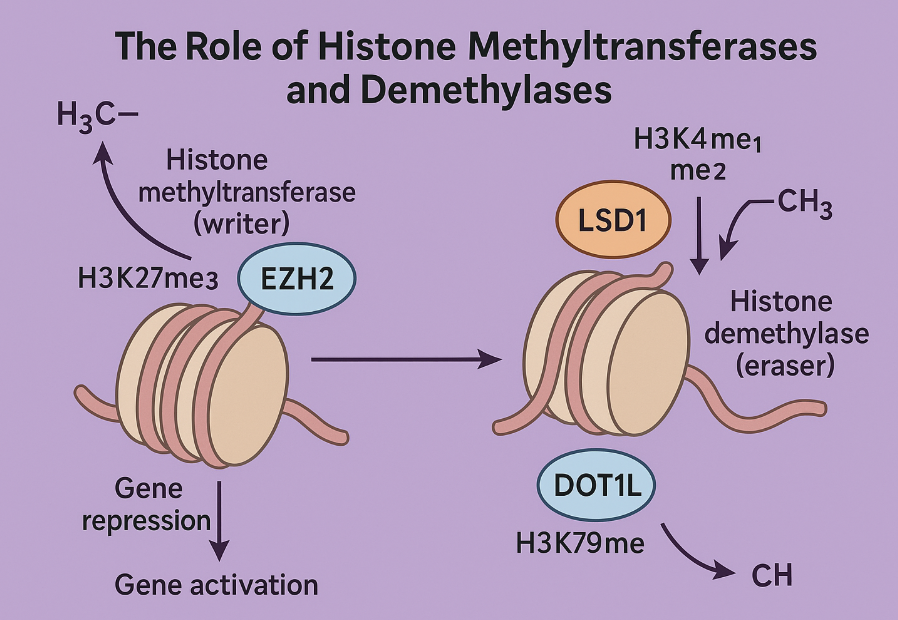
Fig.1 Histone Methylation: EZH2, DOT1L & LSD1 Explained
These enzymes don’t act alone—they function within larger chromatin remodeling complexes, and their effects depend heavily on cellular context. This complexity makes them both challenging and highly promising drug targets. Their selective inhibition offers a path to reactivate silenced genes, suppress oncogenic drivers, and ultimately reshape the cancer epigenome from within.
From Bench to Bedside: Promising Inhibitors in Clinical Trials
Translating epigenetic insights into therapeutic strategies has led to the development of small-molecule inhibitors that specifically target histone-modifying enzymes. Several of these compounds are now in clinical trials, showing encouraging signs of efficacy—especially in hematological malignancies.
DOT1L Inhibitor: Pinometostat (EPZ-5676)
DOT1L catalyzes methylation of H3K79, an activating histone mark. In leukemias with MLL gene rearrangements, fusion proteins aberrantly recruit DOT1L to oncogenic loci like HOXA9, fueling uncontrolled cell growth. Pinometostat, developed by Epizyme, was the first selective DOT1L inhibitor to enter clinical trials. In preclinical studies, it selectively killed MLL-rearranged leukemia cells while sparing normal ones. Despite challenges with oral bioavailability, intravenous administration showed promising anti-leukemic activity and tolerability in early-phase trials.
EZH2 Inhibitors: Tazemetostat and GSK126
EZH2, responsible for placing the repressive H3K27me3 mark, is frequently mutated or overexpressed in cancers such as diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma. Tazemetostat (EPZ-6438), another Epizyme candidate, has demonstrated strong efficacy in EZH2-mutant lymphomas and is now undergoing advanced clinical testing. Similarly, GSK126 from GlaxoSmithKline has shown selective suppression of EZH2-mutant tumors, leading to tumor regression in preclinical models.
LSD1 Inhibitors: ORY-1001 and GSK2879552
LSD1 demethylates H3K4me1/2, repressing gene transcription. In acute myeloid leukemia (AML) and small cell lung cancer (SCLC), LSD1 is overexpressed and sustains a proliferative, stem-like state. ORY-1001, a highly selective LSD1 inhibitor by Oryzon Genomics, has shown strong differentiation-inducing effects in MLL-rearranged leukemia. GSK2879552, another promising compound, is in clinical trials for AML and SCLC, demonstrating antiproliferative effects in preclinical studies.
These inhibitors are not just blocking enzymes—they are rewiring the cancer cell’s epigenetic memory. Their emergence signals a shift from broad-spectrum cytotoxic drugs toward targeted, mechanism-based therapies.
Challenges on the Road: Selectivity, Delivery, and Resistance
Despite the excitement surrounding epigenetic therapies, several challenges remain on the road to successful clinical implementation. Developing inhibitors that precisely target histone methyltransferases and demethylases requires navigating a landscape of structural similarity, metabolic complexity, and adaptive cancer behavior.
Selectivity and Off-Target Effects
One of the biggest hurdles in targeting histone-modifying enzymes is selectivity. Many lysine methyltransferases and demethylases share conserved domains—such as the SET domain in KMTs or the Jumonji C domain in KDMs—making it difficult to design inhibitors that are both potent and specific. For example, LSD1 shares enzymatic homology with monoamine oxidases (MAO-A and MAO-B), which are critical for neurotransmitter metabolism. As a result, early LSD1 inhibitors like tranylcypromine exhibited undesirable side effects like dizziness and hypertension due to non-selective MAO inhibition.
Drug Delivery and Bioavailability
Another major issue is pharmacokinetics. Several promising compounds, including DOT1L inhibitor Pinometostat (EPZ-5676), suffer from poor oral bioavailability, necessitating continuous intravenous infusion. This complicates treatment logistics, increases costs, and limits patient access—especially in outpatient or low-resource settings. Enhancing cell permeability and metabolic stability while preserving target affinity is a top priority for next-generation epigenetic drugs.
Resistance and Tumor Heterogeneity
As with most targeted therapies, drug resistance can emerge in response to prolonged treatment. Tumor cells may compensate by activating alternative epigenetic pathways, upregulating redundant enzymes, or altering the chromatin landscape to maintain oncogenic programs. Additionally, tumor heterogeneity—the coexistence of multiple cellular subtypes within a tumor—means that only a fraction of cells may be sensitive to a given inhibitor, leading to relapse or incomplete response.
Overcoming these challenges will require smarter drug designs, combination therapies, and personalized treatment strategies that incorporate molecular profiling. The field is evolving rapidly, but thoughtful navigation of these obstacles is essential to translate epigenetic therapies into durable clinical benefits.
Future Directions: Beyond Inhibitors to Epigenetic Synergy
As the field of epigenetic therapy matures, researchers are looking beyond single-enzyme inhibitors to explore synergistic strategies and novel target classes that could unlock even more effective cancer treatments. While targeting histone methyltransferases and demethylases has already shown clinical promise, the next frontier lies in expanding and integrating these tools within broader therapeutic frameworks.
Combination Therapies for Greater Efficacy
One emerging approach is combining histone methylation inhibitors with histone deacetylase (HDAC) inhibitors, DNA methyltransferase (DNMT) inhibitors, or chemotherapeutic agents. These combinations aim to attack cancer cells on multiple epigenetic fronts, disrupting redundant survival pathways and reactivating silenced tumor suppressor genes. For example, combining LSD1 inhibitors with HDAC inhibitors such as panobinostat has shown synergistic lethality in acute myeloid leukemia (AML) models, leading to enhanced cell death compared to either agent alone.
Targeting the Readers of Methylation
Beyond the writers (methyltransferases) and erasers (demethylases), a new wave of interest is focusing on the readers—proteins that recognize and bind to specific methylation marks. These include domains like bromodomains, Tudor, and chromodomains, which help recruit chromatin-remodeling complexes and influence transcription. While inhibitors of bromodomains (like BET inhibitors) are already in clinical development, small-molecule antagonists of methylation readers remain underexplored. Targeting these proteins could add a new dimension to the precision control of gene expression.
Personalization and Epigenomic Profiling
Future therapies will likely rely on epigenomic profiling to stratify patients based on their chromatin signatures. As high-throughput sequencing and biomarker discovery improve, clinicians could match patients with the epigenetic therapies most likely to restore normal gene expression patterns in their specific cancer subtype.
In summary, the future of cancer epigenetics lies not just in better inhibitors, but in integrative, targeted, and personalized interventions. The blueprint for the next generation of anticancer strategies is being written—one histone mark at a time.
References:
Jones, B., Su, H., Bhat, A., Lei, H., Bajko, J., Hevi, S., et al. (2008). The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genetics, 4(6), e1000190.
https://doi.org/10.1371/journal.pgen.1000190
McGrath, J., & Trojer, P. (2015). Targeting histone lysine methylation in cancer. Pharmacology & Therapeutics, 150, 1–22.
https://doi.org/10.1016/j.pharmthera.2015.01.003
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell, 119(7), 941–953.
https://doi.org/10.1016/j.cell.2004.12.012
Morera, L., Lübbert, M., & Jung, M. (2016). Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clinical Epigenetics, 8(57).
https://doi.org/10.1186/s13148-016-0223-4
McCabe, M. T., et al. (2012). EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature, 492(7427), 108–112.
https://doi.org/10.1038/nature11606
Nguyen, A. T., & Zhang, Y. (2011). The diverse functions of Dot1 and H3K79 methylation. Genes & Development, 25(13), 1345–1358.
https://doi.org/10.1101/gad.2057811
Daigle, S. R., Olhava, E. J., Therkelsen, C. A., et al. (2011). Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell, 20(1), 53–65.
https://doi.org/10.1016/j.ccr.2011.06.009
Knutson, S. K., Kawano, S., Minoshima, Y., et al. (2014). Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Molecular Cancer Therapeutics, 13(4), 842–854.
https://doi.org/10.1158/1535-7163.MCT-13-0773
Schenk, T., Chen, W. C., Göllner, S., et al. (2012). Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nature Medicine, 18(4), 605–611.
https://doi.org/10.1038/nm.2661
Binda, C., Valente, S., Romanenghi, M., et al. (2010). Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. Journal of the American Chemical Society, 132(19), 6827–6833.
https://doi.org/10.1021/ja100039t
Fiskus, W., Sharma, S., Shah, B., et al. (2014). Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia, 28(11), 2155–2164.
https://doi.org/10.1038/leu.2014.169
Wagner, T., Robaa, D., Sippl, W., & Jung, M. (2014). Mind the methyl: methyllysine binding proteins in epigenetic regulation. ChemMedChem, 9(3), 466–483.