DNA Methylation Reimagined: From Epigenetic Stability to Human Disease
Abstract
DNA methylation has long been portrayed as a process of sweeping erasure and re-establishment across the genome. However, recent high-resolution studies reveal a more nuanced picture in which stability and change are carefully compartmentalized. CpG-island promoters, critical for gene regulation, remain largely unmethylated and protected, while repetitive elements and intergenic regions experience the majority of methylation dynamics. This refined view explains how developmental reprogramming preserves essential regulatory circuits, while transposon silencing and germline-specific processes introduce necessary variability. Central to this balance are DNA methyltransferases (DNMTs) and their cofactors, which establish, maintain, and safeguard methylation marks. Mutations in DNMT genes lead to distinctive human diseases, including immunodeficiency, overgrowth syndromes, neurological disorders, and various cancers. By connecting molecular mechanisms to clinical outcomes, this framework underscores the importance of precision in epigenetic regulation and highlights the therapeutic potential of targeting methylation pathways.
Introduction – Rethinking DNA Methylation
For decades, DNA methylation has been portrayed as a dramatic process where the genome undergoes sweeping cycles of erasure and rewriting during development. This idea of “global methylation waves” captured the imagination of researchers and the public alike, shaping much of how epigenetics has been explained. However, more precise genome-wide mapping has revealed that this picture is oversimplified. Instead of the entire genome being erased and repatterned, the reality is that methylation stability and change are distributed unevenly across genomic landscapes.
One of the most striking insights from recent research is that CpG-island promoters—regions of DNA near transcription start sites enriched in cytosine–guanine dinucleotides—are remarkably stable. These islands are typically unmethylated and remain so across cell types and developmental stages. In contrast, repetitive elements such as long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), and long terminal repeats (LTRs) are heavily methylated and experience most of the dynamic changes that were once thought to be universal. This compartmentalized view reframes methylation as a selective, rather than global, process.
The significance of this shift cannot be overstated. By recognizing that methylation dynamics largely occur outside of promoters and regulatory CpG islands, researchers gain a clearer picture of how gene expression remains stable despite epigenetic remodeling elsewhere. This stability ensures that essential regulatory circuits governing cell identity are preserved even while the genome undergoes extensive developmental transitions.
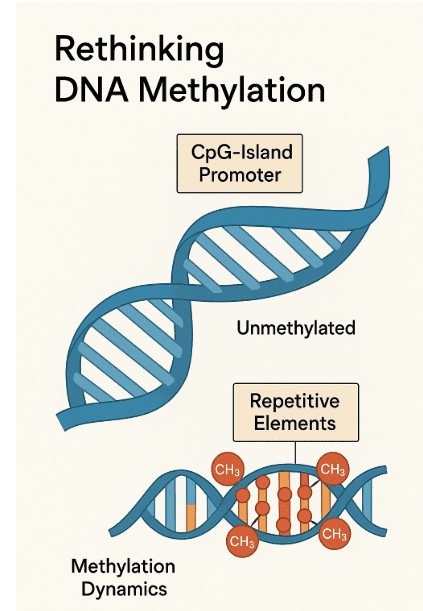
Fig 1 CpG-Island Stability vs. Repetitive Element Methylation
From a disease perspective, this refined model of methylation helps explain why disruptions in DNA methyltransferase enzymes or chromatin-associated factors lead to highly specific pathologies. Rather than being caused by indiscriminate “loss” or “gain” of methylation, disorders are increasingly understood as failures to maintain selective protection or proper targeting in vulnerable genomic compartments.
In short, the contemporary understanding of DNA methylation moves us beyond the metaphor of a genome wiped clean and rewritten. Instead, it reveals a sophisticated system of precision, protection, and compartmentalization, where the majority of methylation dynamics take place in repetitive elements while promoters remain largely safeguarded. This new perspective sets the stage for deeper discussions about how methylation is established, maintained, and linked to human disease.
Where Methylation Lives in the Genome
DNA methylation is not distributed randomly across the genome. Instead, it follows a compartmentalized pattern that reflects both evolutionary history and functional necessity. Understanding where methylation resides is critical, because it helps clarify why some regions of the genome are highly dynamic while others are remarkably stable.
The majority of methylation marks in mammalian genomes are found within repetitive DNA sequences, including transposon-derived elements such as LINEs (long interspersed nuclear elements), SINEs (short interspersed nuclear elements), and LTRs (long terminal repeats). These regions make up a large fraction of the genome, and methylation serves as a powerful silencing mechanism to prevent transposon mobilization. Without this repression, transposons could destabilize the genome by inserting into functional genes or regulatory elements. Thus, heavy methylation of repeats represents a defensive strategy that maintains genomic integrity.
Another major compartment is gene bodies, where DNA methylation often correlates with transcriptional activity. Intragenic methylation does not generally repress gene expression but may fine-tune transcription by preventing spurious initiation within coding sequences. This nuanced role contrasts with the classical view of methylation solely as a silencing mechanism, highlighting its context-dependent function.
By contrast, CpG islands at gene promoters and first exons—although they account for less than 0.5% of the genome—tend to remain unmethylated. Their protection from methylation is vital for maintaining transcriptional competence, particularly for genes involved in cell identity, housekeeping functions, and developmental regulation. Exceptions exist: certain tissue-specific genes and imprinted loci do acquire promoter methylation, leading to long-term silencing. But overall, promoter CpG islands stand out as stable “methylation-free zones” in an otherwise heavily marked genome.
This compartmentalized organization has profound implications. It shows that methylation dynamics are not genome-wide, but localized mainly to repetitive elements and intergenic regions. It also explains why disturbances in methylation machinery often manifest in specific genomic vulnerabilities rather than universal disruption. For example, failures to maintain methylation at repeats can lead to genome instability, while inappropriate targeting of CpG islands can cause aberrant silencing of tumor suppressor genes in cancer.
In sum, the genomic geography of methylation reflects a delicate balance: repression of repeats, modulation of gene bodies, and protection of promoters. Recognizing this balance is essential for interpreting epigenome maps and for understanding how epigenetic dysregulation contributes to disease.
Developmental Dynamics – Stability vs. Change
DNA methylation undergoes dramatic reorganization during mammalian development, but these changes are more selective than once thought. Traditional models described two major “waves” of methylation reprogramming: an erasure phase in early embryogenesis followed by de novo methylation, and a separate cycle in the germline. These waves were initially perceived as genome-wide, yet detailed mapping has revealed that stability and change are compartmentalized.
In early embryos, global demethylation resets much of the paternal and maternal methylation landscape. However, not all regions are equally affected. Imprinting control regions (ICRs) and differentially methylated regions (DMRs), which are essential for parent-of-origin gene expression, resist this erasure and preserve epigenetic memory. Likewise, some classes of transposons—particularly young, CpG-rich retroelements—remain protected from demethylation. This selective resistance highlights the precision of developmental programming.
Following implantation, the genome undergoes extensive de novo methylation to establish tissue-specific epigenomes. Again, this does not occur uniformly. Repetitive elements and intergenic regions are the primary substrates for re-methylation, while CpG-island promoters remain mostly unmethylated. This selective pattern ensures that essential regulatory genes remain poised for activation, while potentially harmful sequences like transposons are repressed.
The germline presents another layer of complexity. Primordial germ cells (PGCs) experience nearly complete erasure of methylation, including imprints, which is essential for resetting the epigenome for the next generation. Once sex determination occurs, male and female germ cells follow distinct trajectories. Male germ cells undergo de novo methylation early and accumulate methylation through numerous mitotic divisions before meiosis. This early acquisition is associated with an increased rate of CpG-to-TpG mutations, reflecting the mutagenic nature of methylated cytosines. Female germ cells, in contrast, remain hypomethylated until later in oocyte growth, showing a delayed but precise re-establishment of methylation.
These dynamics underscore a key point: epigenetic reprogramming is not a blunt erasure-and-rewrite process but a finely tuned balance of protection, erasure, and re-establishment. The genome preserves critical regulatory regions while allowing flexibility elsewhere, ensuring both developmental plasticity and genomic stability. Disruptions in these processes—such as inappropriate erasure or failure to protect key regions—can have profound implications, including developmental disorders, infertility, and tumorigenesis.
The Machinery of Control – DNMTs and Protectors
The establishment and maintenance of DNA methylation patterns depend on a highly coordinated network of enzymes and chromatin regulators. At the center of this network are the DNA methyltransferases (DNMTs), which function either to install new methylation marks (de novo methylation) or to preserve them through cell division (maintenance methylation). Together, they create a balance between flexibility and stability in the epigenome.
De novo methylation is primarily catalyzed by DNMT3A and DNMT3B, with DNMT3L serving as an essential cofactor that stimulates their activity. These enzymes are most active during early development and in the germline, where large-scale methylation patterns must be set anew. Importantly, DNMT3A and DNMT3B do not act indiscriminately; their activity is influenced by chromatin context. For example, they preferentially target regions marked by unmethylated histone H3 lysine 4 (H3K4), avoiding active promoters that must remain accessible for transcription. In male germ cells, components of the piRNA pathway—such as MILI and MIWI2—provide upstream guidance to DNMT3 enzymes, ensuring proper repression of transposons and safeguarding genomic integrity.
On the other hand, maintenance methylation is carried out by DNMT1, which copies methylation patterns from the parental DNA strand to the daughter strand during replication. This process is facilitated by UHRF1, a protein that recognizes hemimethylated CpGs and recruits DNMT1 to the correct sites. Structural studies have revealed that DNMT1 possesses domains such as the replication foci targeting sequence (RFTS) and the CXXC domain, which prevent inappropriate methylation of unmethylated CpG islands and promote high fidelity in copying methylation marks. These safeguards ensure that stable regulatory patterns—such as those at imprinting control regions—are preserved across cell divisions.
In addition to DNMTs, specialized protective mechanisms prevent inappropriate methylation at CpG-island promoters. A key player is FBXL10 (also known as KDM2B), which recruits polycomb repressive complexes (PRC1 and PRC2) to CpG islands and shields them from aberrant de novo methylation. Loss of this protective function has been linked to abnormal promoter hypermethylation, a hallmark of certain pediatric cancers. Thus, while DNMTs establish and maintain methylation, proteins like FBXL10 act as guardians of the unmethylated state, ensuring regulatory genes remain poised for proper expression.
Taken together, these findings reveal that DNA methylation is not merely a chemical modification imposed on DNA but the outcome of a multi-layered system of targeting, copying, and protection. The precision of this machinery is essential for normal development, and its breakdown can lead directly to disease.
From Mechanisms to Medicine – Human Disease Links
The detailed understanding of DNA methylation machinery has illuminated how defects in this system give rise to human disease. Mutations in DNA methyltransferases (DNMTs) and associated regulatory proteins do not simply cause random methylation errors; instead, they produce distinctive clinical syndromes that reflect the precise roles of these enzymes in development and homeostasis.
One of the clearest examples is Immunodeficiency, Centromeric instability, and Facial anomalies (ICF) syndrome, caused by recessive mutations in DNMT3B. Patients with ICF syndrome exhibit hypomethylation at pericentromeric satellite repeats, leading to chromosomal instability, immune dysfunction, and characteristic facial features. This condition demonstrates how DNMT3B’s failure to methylate repetitive DNA undermines genome integrity and immune system development.
Another important case is Tatton–Brown–Rahman syndrome, a rare overgrowth disorder caused by germline mutations in DNMT3A. Affected individuals often present with tall stature, intellectual disability, and distinctive craniofacial features. Beyond germline mutations, somatic mutations in DNMT3A—especially the recurrent R882 hotspot mutation—are among the most common genetic lesions in acute myeloid leukemia (AML) and myelodysplastic syndromes. These mutations reduce DNMT3A’s enzymatic activity and alter methylation patterns in hematopoietic stem cells, driving malignant transformation.
DNMT1 mutations give rise to a strikingly different phenotype: progressive adult-onset neurological syndromes characterized by combinations of ataxia, sensorineural deafness, narcolepsy, and dementia. These disorders, often referred to collectively as hereditary sensory and autonomic neuropathies with dementia, highlight DNMT1’s central role in maintenance methylation. Mutations typically cluster in the replication foci targeting sequence (RFTS) domain, disrupting the enzyme’s ability to accurately copy methylation marks during DNA replication.
While DNMT3L has no known human disease associations to date, studies in mouse models suggest that its absence leads to infertility and embryonic lethality. This predicts that pathogenic DNMT3L variants in humans may be incompatible with life or fertility, explaining the lack of identified patients.
The clinical relevance of these discoveries extends to cancer biology. Aberrant methylation patterns, such as hypermethylation of CpG-island promoters, are now recognized as hallmarks of tumorigenesis. Therapies targeting epigenetic regulators, including DNMT inhibitors like azacitidine and decitabine, are already in clinical use for hematological malignancies. Thus, research into DNMT function bridges molecular mechanisms with therapeutic innovation, offering new hope for patients affected by both rare syndromes and common cancers.
References
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes & Development, 16(1), 6–21.
https://doi.org/10.1101/gad.947102
Smith, Z. D., & Meissner, A. (2013). DNA methylation: Roles in mammalian development. Nature Reviews Genetics, 14(3), 204–220.
https://doi.org/10.1038/nrg3354
Smith, Z. D., Chan, M. M., Mikkelsen, T. S., Gu, H., Gnirke, A., Regev, A., & Meissner, A. (2012). A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature, 484(7394), 339–344.
https://doi.org/10.1038/nature10960
Greenberg, M. V. C., & Bourc’his, D. (2019). The diverse roles of DNA methylation in mammalian development and disease. Nature Reviews Molecular Cell Biology, 20(10), 590–607.
https://doi.org/10.1038/s41580-019-0159-6
Deaton, A. M., & Bird, A. (2011). CpG islands and the regulation of transcription. Genes & Development, 25(10), 1010–1022.
https://doi.org/10.1101/gad.2037511
Jones, P. A. (2012). Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nature Reviews Genetics, 13(7), 484–492.
https://doi.org/10.1038/nrg3230
Slotkin, R. K., & Martienssen, R. (2007). Transposable elements and the epigenetic regulation of the genome. Nature Reviews Genetics, 8(4), 272–285.
https://doi.org/10.1038/nrg2072
Lister, R., Pelizzola, M., Dowen, R. H., Hawkins, R. D., Hon, G., Tonti-Filippini, J., … Ecker, J. R. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature, 462(7271), 315–322.
https://doi.org/10.1038/nature08514
Guibert, S., Forné, T., & Weber, M. (2012). Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Research, 22(4), 633–641.
https://doi.org/10.1101/gr.130997.111
Seisenberger, S., Peat, J. R., & Reik, W. (2013). Conceptual links between DNA methylation reprogramming in the early embryo and primordial germ cells. Current Opinion in Cell Biology, 25(3), 281–288.
https://doi.org/10.1016/j.ceb.2013.02.013
Smallwood, S. A., & Kelsey, G. (2012). De novo DNA methylation: A germ cell perspective. Trends in Genetics, 28(1), 33–42.
https://doi.org/10.1016/j.tig.2011.09.004
Jeltsch, A., & Jurkowska, R. Z. (2016). Allosteric control of mammalian DNA methyltransferases: A new regulatory paradigm. Nucleic Acids Research, 44(18), 8556–8575.
https://doi.org/10.1093/nar/gkw723
Bestor, T. H. (2015). The DNA methyltransferases of mammals. Cold Spring Harbor Perspectives in Biology, 7(2), a019497.
https://doi.org/10.1101/cshperspect.a019497
Tatton-Brown, K., & Rahman, N. (2013). The NSD1 and DNMT3A overgrowth syndromes. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 163(2), 86–91.