Decoding DNA Methylation: What We Now Know About Epigenetics and Development
Abstract
DNA methylation is a cornerstone of epigenetic regulation, influencing gene expression, genomic stability, and developmental programming. While early models suggested widespread and dynamic methylation changes across the genome, recent high-resolution analyses have challenged this view. Drawing on insights from Edwards et al. (2017), this blog post revisits the landscape of DNA methylation, revealing that functionally significant methylation is confined to a small fraction of the genome—primarily CpG island promoters, imprinting control regions, and transposable elements. We explore how methylation patterns are established and maintained by a suite of specialized enzymes, the DNMTs, and examine how disruptions in these processes can lead to diverse human diseases, from immunodeficiency syndromes to neurological and hematological disorders. By understanding where methylation matters—and where it doesn’t—we gain a clearer picture of its true role in biology and disease.
Introduction: The Epigenetic Blueprint
Why do genetically identical twins sometimes develop different diseases? How does a liver cell know not to become a neuron, even though it has the same DNA? The answer lies beyond the genetic code—in the dynamic world of epigenetics.
Among the most studied epigenetic mechanisms is DNA methylation, a process that adds methyl groups to specific cytosines, typically at CpG dinucleotides. These chemical tags don’t change the DNA sequence but influence how genes are turned on or off. DNA methylation is essential for a range of biological processes, including embryonic development, genomic imprinting, X-chromosome inactivation, and suppression of transposable elements.
Despite its fundamental role, our understanding of DNA methylation has evolved dramatically. Early models from the 1980s painted a picture of sweeping methylation changes across the genome during development. These views emerged at a time when the genome was poorly annotated, and assumptions about function were based on limited data.
Fast forward to today, and whole-genome methylation profiling has brought a sharper lens to this field. A landmark 2017 review by Edwards and colleagues challenges many outdated assumptions and offers a more nuanced understanding. They demonstrate that only a small fraction of the genome—less than 10%—shows evidence of biologically meaningful methylation patterns. Most changes during development occur in non-functional regions, such as old transposons or repetitive DNA, rather than in gene promoters or enhancers.
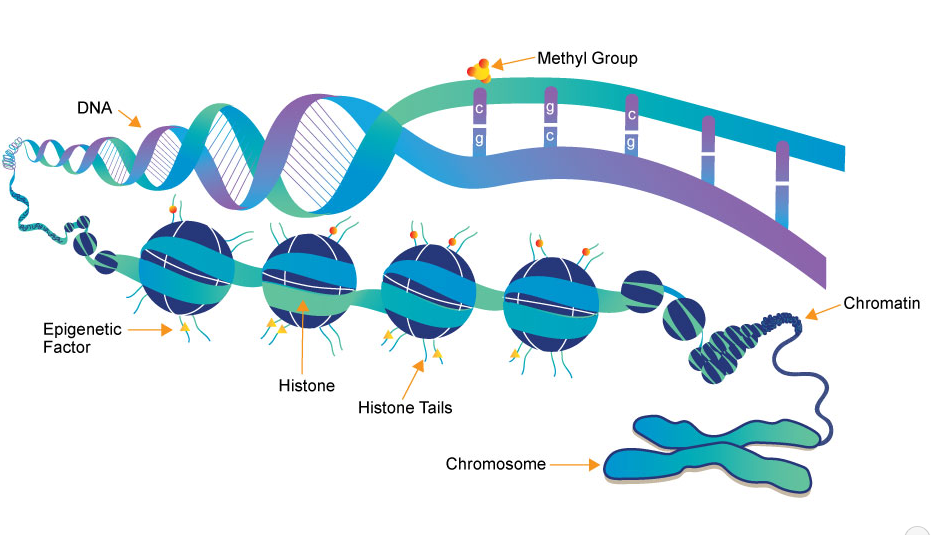
Fig.1 Representation of the chromatin structure, including histones and DNA, which become available to epigenetic marks.
In this blog series, we’ll unpack the findings from Edwards et al., shedding light on how DNA methylation really works, what regulates it, and what happens when these processes break down. Understanding this revised blueprint is key to grasping both normal development and the origins of many epigenetic diseases.
Rethinking the Role of Methylation in the Human Genome
For years, scientists believed that DNA methylation patterns across the genome played widespread regulatory roles—silencing genes, guiding development, and shaping cellular identity across the entire genome. However, high-resolution methylome data from recent studies tell a different story: much of DNA methylation may actually be biologically irrelevant.
According to Edwards et al. (2017), only about 10% of the mammalian genome shows signs of functional constraint or selective pressure. The rest—consisting largely of repetitive sequences, inactive transposons, satellite DNA, and unannotated regions—is evolving neutrally. Not surprisingly, these areas are also where most methylation changes occur during development. That suggests methylation is acting more like epigenetic background noise in many parts of the genome.
This shifts our focus to the areas where methylation truly matters. CpG islands, which are short, G+C-rich stretches often found in gene promoters and first exons, stand out. These regions are typically unmethylated, allowing transcription to proceed. Interestingly, Edwards et al. highlight that less than 0.5% of the genome consists of these unmethylated CpG-rich regulatory regions. In contrast, transposon-derived sequences, such as SINEs, LINEs, and LTRs, are densely methylated but don’t generally regulate genes. Their methylation serves more of a genomic defense role—silencing potential mobile elements to maintain genome integrity.
These findings not only redefine the biological importance of methylation but also underscore the limitations of early epigenetic models, which lacked a genome-wide perspective. The distinction between functional and non-functional methylation has major implications for epigenetic research, biomarker discovery, and understanding diseases like cancer, where methylation changes are often observed but not always biologically relevant.
The genome is not a uniform canvas; it’s a mosaic of functional compartments, and DNA methylation reflects this complexity. Understanding where methylation counts—and where it doesn’t—is the first step toward truly decoding the epigenome.
Methylation in Development
For decades, developmental biologists have taught that mammalian embryos undergo two dramatic “waves” of DNA methylation changes: one during the formation of germ cells and another shortly after fertilization. These processes—demethylation followed by remethylation—were believed to reset the epigenome across nearly the entire genome. But recent research reveals a more complex and selective reality.
Edwards et al. (2017) clarify that these methylation waves affect only specific portions of the genome, primarily regions with low regulatory importance. While the majority of the genome does undergo demethylation during germline and early embryonic stages, critical regulatory regions like CpG island promoters and imprinting control regions (ICRs) often escape these changes. These sequences remain unmethylated or are protected to preserve epigenetic inheritance and gene regulation fidelity.
One key revelation is that young, CpG-rich transposons—which can potentially disrupt genome stability—also resist demethylation, remaining silenced during early development. Meanwhile, sex-specific differences emerge in how methylation patterns are established and maintained. In males, de novo methylation occurs around birth and is maintained throughout spermatogenesis. In contrast, female germ cells acquire methylation later, during oocyte maturation, and undergo fewer mitotic divisions, leading to fewer opportunities for mutational errors such as C→T transitions.
This sexual dimorphism has evolutionary and medical implications. For example, paternal alleles are more frequently associated with CpG-related mutations, contributing to certain sporadic genetic disorders.
In essence, the once-popular “double-dip” model oversimplifies the methylation story. The genome is not methylated or demethylated as a monolith. Instead, it undergoes selective, sequence-specific changes, preserving methylation in regions essential for development while resetting others that are evolutionarily neutral. Recognizing these exceptions helps refine our understanding of epigenetic reprogramming and lays the groundwork for interpreting methylation-related disease mechanisms.
The Machinery Behind the Marks: DNMTs and Their Precision Tools
Behind the seemingly simple act of DNA methylation lies a sophisticated and highly coordinated enzymatic system. At the core of this system are the DNA methyltransferases (DNMTs)—a family of enzymes responsible for establishing and maintaining methylation patterns across the genome. Each plays a distinct role in epigenetic regulation, with finely tuned mechanisms that ensure both specificity and stability.
DNMT3A and DNMT3B are known as de novo methyltransferases. They establish new methylation patterns during early development and in germ cells. Their activity is directed to specific genomic regions, often guided by histone modifications and accessory proteins. One such protein is DNMT3L, which doesn’t methylate DNA itself but forms complexes with DNMT3A and DNMT3B. It recognizes nucleosomes marked by unmethylated H3K4, a signal associated with regions primed for methylation.
On the other hand, DNMT1 is the principal enzyme for maintenance methylation. After DNA replication, DNMT1 restores methylation on the newly synthesized strand, preserving the methylation status of the parent strand. This ensures that epigenetic memory is faithfully passed on through cell divisions. Its preference for hemimethylated DNA is reinforced by its interaction with UHRF1, a multidomain protein that binds to hemimethylated CpG sites and recruits DNMT1 to replication foci.
Structurally, DNMT1 features a CXXC domain that binds unmethylated CpGs, thereby preventing inappropriate methylation. It also contains RFTS and BAH domains that regulate its activity through autoinhibition and potential histone interactions.
Interestingly, while CpG island promoters are generally protected from methylation, certain proteins like FBXL10 (also known as KDM2B) help maintain this protection. FBXL10 counteracts the tendency of polycomb repressive complexes (PRC1/2) to promote methylation at specific loci. This regulatory balance is crucial; when disrupted, it can lead to gene silencing and contribute to diseases like cancer.
Together, the DNMT machinery exemplifies precision in epigenetic control—targeting, timing, and maintaining methylation with remarkable specificity.
When Methylation Goes Wrong: Human Disorders Linked to DNMT Mutations
While DNA methylation is essential for normal development and genome stability, its disruption can lead to a range of human diseases. Mutations in DNA methyltransferase (DNMT) genes—key enzymes responsible for establishing and maintaining methylation—have been directly linked to several distinct genetic disorders. These conditions demonstrate how finely tuned the epigenetic machinery must be to sustain health.
DNMT3B, one of the enzymes responsible for de novo methylation, is mutated in ICF syndrome type 1 (Immunodeficiency, Centromeric instability, and Facial anomalies syndrome). This rare, autosomal recessive disorder is characterized by severe immunodeficiency, chromosome instability, and facial dysmorphisms. Patients exhibit widespread hypomethylation, particularly in satellite DNA, which leads to chromosomal abnormalities. Interestingly, although hypomethylation is extensive, only certain cell types exhibit instability—highlighting the complexity of epigenetic dysfunction.
DNMT3A mutations are linked to two major disease types. Germline mutations in DNMT3A cause Tatton–Brown–Rahman syndrome, a congenital overgrowth disorder involving tall stature, intellectual disabilities, and characteristic facial features. On the other hand, somatic mutations—especially at codon R882—are found in approximately 15% of acute myeloid leukemia (AML) cases. These mutations alter methylation dynamics and may contribute to the initiation and progression of leukemia, though the exact methylation changes driving malignancy are still under investigation.
DNMT1, the maintenance methyltransferase, is mutated in rare adult-onset neurological syndromes. These include cerebellar ataxia, deafness, narcolepsy, and dementia. All known mutations cluster within the replication focus targeting sequence (RFTS) of DNMT1, disrupting its interaction with chromatin and potentially causing toxic protein aggregation in neurons. Curiously, these conditions affect the nervous system despite the fact that mature neurons are post-mitotic and perform little DNA replication, suggesting mechanisms beyond classical methylation may be at play.
These diverse disorders underscore the critical roles of DNMTs in both development and disease. Even subtle disruptions to the methylation machinery can result in complex, tissue-specific phenotypes—emphasizing the importance of precision in epigenetic regulation.
References
Edwards, J. R., Yarychkivska, O., Boulard, M., & Bestor, T. H. (2017). DNA methylation and DNA methyltransferases. Epigenetics & Chromatin, 10(23).
https://doi.org/10.1186/s13072-017-0130-8
Lister, R., Pelizzola, M., Dowen, R. H., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature, 462(7271), 315–322.
https://doi.org/10.1038/nature08514
Bestor, T. H., Edwards, J. R., & Boulard, M. (2015). Notes on the role of dynamic DNA methylation in mammalian development. PNAS, 112(22), 6796–6799.
https://doi.org/10.1073/pnas.1415301112
Ioshikhes, I. P., & Zhang, M. Q. (2000). Large-scale human promoter mapping using CpG islands. Nature Genetics, 26(1), 61–63.
Eddy, S. R. (2013). The ENCODE project: missteps overshadowing a success. Current Biology, 23(7), R259–R261.
https://doi.org/10.1016/j.cub.2013.02.041
Seisenberger, S., Andrews, S., Krueger, F., et al. (2012). The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Molecular Cell, 48(6), 849–862.
https://doi.org/10.1016/j.molcel.2012.11.001
Bourc’his, D., & Bestor, T. H. (2004). Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature, 431(7004), 96–99.
https://doi.org/10.1038/nature02886
Ooi, S. K. T., Qiu, C., Bernstein, E., et al. (2007). DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature, 448(7154), 714–717.
https://doi.org/10.1038/nature05987
Baets, J., Deconinck, T., Smets, K., et al. (2015). Defects of mutant DNMT1 are linked to a spectrum of neurological disorders. Brain, 138(4), 845–861.