Decoding Cancer Pathways: The Multifaceted Role of PKC Isozymes in Tumor Progression and Therapy
Abstract
Protein Kinase C (PKC) isozymes are a diverse family of serine/threonine kinases that play critical roles in regulating cellular signaling pathways. Their isoform-specific functions in processes such as proliferation, apoptosis, migration, and differentiation make them central to the molecular mechanisms of cancer. This blog explores the classification of PKC isozymes and their context-dependent involvement in various cancers, from breast and colon cancer to gliomas. It also highlights their emerging value as diagnostic markers and therapeutic targets, particularly in the regulation of cancer stem cells and multidrug resistance. A deeper understanding of PKC signaling may open new avenues for precision oncology and combination therapies in the future.
Introduction – Why Protein Kinase C (PKC) Matters in Cancer
Cancer is a complex and adaptive disease driven by disruptions in the regulatory pathways that govern normal cell growth, differentiation, and death. Among the numerous molecules involved in these processes, Protein Kinase C (PKC) stands out as a key player in orchestrating cellular responses to external and internal stimuli. PKC refers to a family of phospholipid-dependent serine/threonine kinases, which are central to many signal transduction pathways across a range of cell types.
PKC isozymes are broadly classified into three major subfamilies based on their activation requirements and structural features:
Conventional (cPKCs) – including PKCα, βI, βII, and γ, which require calcium, diacylglycerol (DAG), and phosphatidylserine.
Novel (nPKCs) – such as PKCδ, ε, η, and θ, which are calcium-independent but still activated by DAG and phosphatidylserine.
Atypical (aPKCs) – including PKCζ and PKCι/λ, which are independent of both calcium and DAG.
The dysregulation of PKC activity—either through overexpression, suppression, or mutation—can tip the balance toward oncogenesis. PKCs are known to influence nearly all hallmarks of cancer, including cell proliferation, evasion of apoptosis, angiogenesis, invasion, and metastasis. For example, overactivation of PKCα or PKCε can promote resistance to apoptosis and enhance metastatic potential in several tumor types.
What makes PKCs particularly intriguing is their context-dependent function. The same isozyme might promote survival in one cancer type and induce apoptosis in another. This duality underscores the importance of understanding the isozyme-specific roles of PKC in the tumor microenvironment.
Given their central role in cancer biology, PKC isozymes are increasingly being explored as therapeutic targets and diagnostic markers, offering a promising frontier for precision oncology.
How PKC Isozymes Drive Cancer Development
The transition from a normal cell to a malignant one involves a cascade of genetic and epigenetic alterations that disrupt intracellular signaling pathways. Among the key regulators of these pathways are Protein Kinase C (PKC) isozymes, which serve as molecular switches in cancer cell signaling. Their activation or inhibition profoundly affects processes such as proliferation, survival, migration, apoptosis, and angiogenesis.
PKC isozymes modulate several oncogenic signaling cascades. Notably, they can activate the Ras/Raf/MEK/ERK pathway, which promotes cell cycle progression and proliferation, as well as the PI3K/Akt/mTOR pathway, which supports metabolic adaptation, cell survival, and resistance to apoptosis. These pathways are frequently upregulated in various tumors and are critical for sustaining the rapid growth and survival of cancer cells.
In contrast, PKCs can also suppress tumor-suppressive signals. For instance, some isozymes downregulate caspase activation, inhibit the Bax subfamily of proapoptotic proteins, or impair p53-mediated transcriptional activity, leading to reduced apoptotic sensitivity. Interestingly, the effects of individual PKC isozymes are highly context-specific. For example, PKCδ acts as an antiapoptotic factor in chronic lymphocytic leukemia but promotes apoptosis in acute myeloid leukemia, highlighting the nuanced roles of PKCs depending on the cellular environment.
Moreover, prolonged exposure to agents like 12-O-tetradecanoylphorbol-13-acetate (TPA) can differentially regulate PKC function. While short-term TPA treatment activates PKCs, long-term exposure leads to their downregulation. This dynamic regulation further complicates therapeutic targeting strategies.
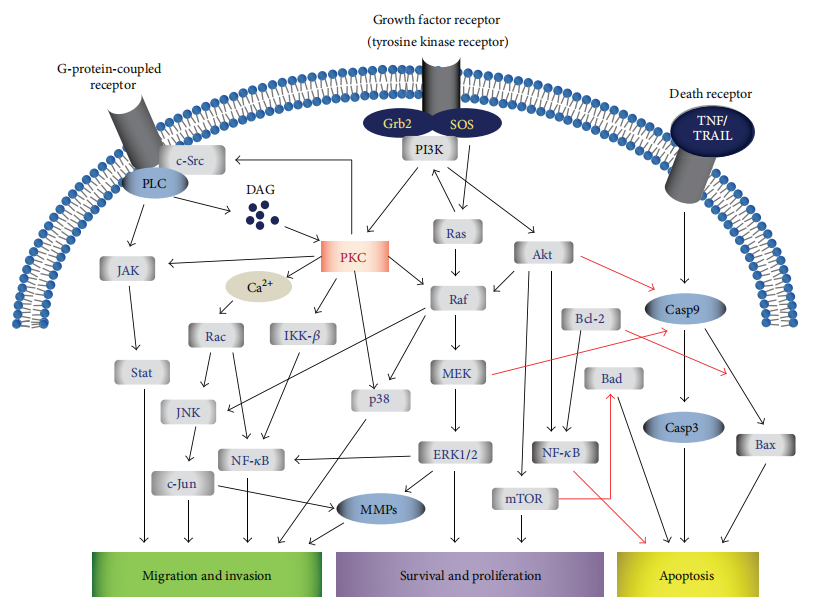
Figure 1: Multiple signaling pathways, involving PKC isozyme regulation and signal transduction, are affected during cancer.
The diverse and sometimes contradictory functions of PKC isozymes underscore the importance of isoform-specific targeting in cancer therapy. Understanding the activation status and downstream effectors of PKC in a given tumor type is critical for designing effective therapeutic interventions.
Cancer-Specific Roles – From Bladder to Brain Tumors
One of the most compelling aspects of Protein Kinase C (PKC) biology in cancer is the isozyme-specific and tumor-specific diversity in their roles. Each PKC isozyme can act either as a promoter or suppressor of cancer progression, depending on the tissue context and tumor microenvironment. This versatility positions PKC isozymes as both biomarkers and therapeutic targets across a wide spectrum of malignancies.
In bladder cancer, PKCα expression is correlated with increased tumor aggressiveness and resistance to the chemotherapy drug adriamycin. In contrast, PKCβI, βII, δ, and η tend to decrease as tumor grade increases, suggesting a suppressive or early-stage role for these isozymes.
Breast cancer presents one of the most complex PKC profiles. PKCα contributes to proliferation, metastasis, and antiestrogen resistance, particularly in ER-negative and triple-negative subtypes. PKCε and PKCη are also linked to antiapoptotic signaling and enhanced survival, while PKCδ displays dual functions—promoting both survival and apoptosis depending on the cellular conditions.
In colon cancer, PKCα appears to function largely as a tumor suppressor by inhibiting Wnt/β-catenin signaling and promoting differentiation. However, PKCβII has been implicated in tumorigenesis and invasion, especially in genetically modified models that overexpress this isozyme.
Gliomas, particularly high-grade glioblastomas, exhibit elevated levels of PKCα, ε, ζ, and ι. These isozymes promote glioma cell proliferation, survival, migration, and invasion. PKCζ, for instance, enhances radiation resistance through interaction with β1-integrin, while PKCι regulates glioma stem cell expansion and invasion via Rac1 and ERK pathways.
This tumor-type-specific functional landscape highlights the diagnostic and therapeutic potential of PKC isozymes. Rather than being universally oncogenic or tumor-suppressive, their behavior is nuanced, reinforcing the need for precision oncology approaches that take isoform expression and activity into account for each cancer type.
PKC Isozymes as Diagnostic and Therapeutic Targets
As research into the molecular underpinnings of cancer advances, Protein Kinase C (PKC) isozymes are increasingly recognized not only as participants in tumor progression but also as actionable biomarkers and drug targets. Because specific PKC isoforms are overexpressed, mutated, or hyperactivated in certain cancers, they hold great promise for both diagnosis and personalized therapy.
From a diagnostic perspective, immunohistochemical detection of PKC expression in tumor tissues is gaining ground. For instance, PKCθ is considered a highly specific diagnostic biomarker for gastrointestinal stromal tumors (GISTs), particularly in cases where other conventional markers like KIT and DOG1 are absent. Similarly, PKCι expression is elevated in non-small cell lung cancer (NSCLC) and ovarian cancer, correlating with aggressive tumor behavior and poor prognosis. High levels of PKCα and PKCβII are also associated with certain breast cancers and diffuse large B-cell lymphoma (DLBCL), respectively, where they serve as indicators of poor therapeutic response and shortened survival.
Therapeutically, targeting PKC isozymes has yielded mixed results. Several broad and isozyme-specific inhibitors have entered preclinical or clinical evaluation. For example, enzastaurin, a selective PKCβ inhibitor, showed promise in reducing proliferation and angiogenesis in preclinical cancer models. However, its success in clinical trials, particularly for lymphoma and glioblastoma, has been limited. Another example is aprinocarsen, an antisense oligonucleotide targeting PKCα mRNA, which has been tested in combination with standard chemotherapeutics for lung, prostate, and colorectal cancers. Despite initial enthusiasm, phase II and III trials have not demonstrated significant clinical benefit.
These outcomes highlight a key challenge: the context-dependent roles of PKC isoforms in different cancers, sometimes acting as oncogenes, other times as tumor suppressors. Therefore, precision targeting strategies—guided by PKC isozyme expression profiles and functional roles—will be essential to realize their full therapeutic potential.
The Future of PKC Research in Cancer Stem Cells and Drug Resistance
As cancer therapy continues to evolve, a persistent challenge remains: how to overcome tumor recurrence, metastasis, and multidrug resistance (MDR). Recent studies have uncovered a pivotal role for Protein Kinase C (PKC) isozymes in these processes, particularly in their regulation of cancer stem cells (CSCs) and their contribution to chemoresistance.
CSCs are a subpopulation of tumor cells with the ability to self-renew, differentiate, and initiate tumor regrowth after therapy. Emerging evidence suggests that specific PKC isozymes, especially PKCι and PKCζ, play crucial roles in sustaining CSC phenotypes. For example, PKCι promotes the expansion and invasion of glioma stem cells via activation of the ERK and Rac1 pathways, thereby enhancing tumorigenic potential and therapeutic resistance. Similarly, PKCε has been implicated in the survival and proliferation of CSCs in breast and prostate cancers by modulating pathways like NF-κB and Akt.
In parallel, PKC isozymes are also directly linked to multidrug resistance mechanisms. Several PKCs, particularly PKCα and PKCθ, regulate the expression and activity of P-glycoprotein (P-gp)—a membrane transporter that actively effluxes chemotherapy drugs out of cancer cells, reducing their efficacy. Through phosphorylation of P-gp and transcriptional control of its gene (MDR1), PKCs help cancer cells adapt and survive under pharmacologic pressure.
These insights highlight the potential of PKC-targeted strategies to sensitize tumors to chemotherapy and reduce recurrence. However, the complexity of PKC signaling demands precision approaches. Therapeutics must be carefully tailored to target the right isozyme in the right cancer context, ideally in combination with agents that disrupt CSC niches or block compensatory pathways.
As the field advances, integrating PKC isozyme profiling into cancer diagnostics and combining PKC inhibitors with immunotherapy or targeted drugs may pave the way for more durable responses and improved patient outcomes.
References
Kang, J.-H. (2014). Protein kinase C (PKC) isozymes and cancer. New Journal of Science, 2014, 1–36.
https://doi.org/10.1155/2014/231418
Steinberg, S. F. (2008). Structural basis of protein kinase C isoform function. Physiological Reviews, 88(4), 1341–1378.
https://doi.org/10.1152/physrev.00034.2007
Griner, E. M., & Kazanietz, M. G. (2007). Protein kinase C and other diacylglycerol effectors in cancer. Nature Reviews Cancer, 7(4), 281–294.
https://doi.org/10.1038/nrc2110
Mackay, H. J., & Twelves, C. J. (2007). Targeting the protein kinase C family: Are we there yet? Nature Reviews Cancer, 7(7), 554–562.
https://doi.org/10.1038/nrc2182
Jackson, D. N., & Foster, D. A. (2004). The enigmatic protein kinase Cδ: complex roles in cell proliferation and survival. FASEB Journal, 18(6), 627–636.
https://doi.org/10.1096/fj.03-0838rev
Martiny-Baron, G., & Fabbro, D. (2007). Classical PKC isoforms in cancer. Pharmacological Research, 55(6), 477–486.