Beyond VEGF: Rethinking Tumor Vasculature to Overcome Resistance in Cancer Therapy
Abstract
Anti-VEGF therapies have revolutionized cancer treatment by targeting tumor angiogenesis, yet their clinical effectiveness has often failed to meet expectations. While early-stage tumors in mouse models respond well to VEGF inhibition, human cancers typically display resistance due to vascular heterogeneity and the emergence of VEGF-independent blood vessels. This blog explores the limitations of current antiangiogenic strategies, highlighting the structural complexity of tumor vasculature and the critical timing of intervention. By examining experimental findings from VEGF-A164 mouse models and considering alternative vascular targets—such as feeder arteries and draining veins—we outline a new direction for anti-vascular cancer therapy. A shift toward multi-targeted and vessel-specific approaches may offer more durable outcomes and reshape the future of oncologic drug development.
The Birth of Antiangiogenic Therapy
The modern era of anti-cancer drug discovery owes much to the visionary work of Dr. Judah Folkman, who, in the early 1970s, proposed a radical idea: that tumors cannot grow beyond a minimal size without establishing a blood supply. This concept, known as tumor angiogenesis, reshaped cancer biology and opened new therapeutic avenues. Folkman further hypothesized that tumors secrete a specific tumor angiogenesis factor (TAF) and that blocking this factor could starve tumors of nutrients and oxygen, effectively halting their growth.
The eventual identification of vascular endothelial growth factor (VEGF-A) as the primary TAF validated Folkman’s hypothesis. VEGF-A is now widely recognized as the master regulator of pathological angiogenesis in cancer. It acts on endothelial cells to stimulate vascular permeability, cell proliferation, and the formation of new blood vessels. Elevated VEGF-A levels have been detected in nearly all solid tumors, reinforcing its role as a critical driver of malignancy.
This discovery led to the development of a new class of antiangiogenic drugs, designed to inhibit VEGF-A or its receptors (VEGFRs). Among the most well-known is bevacizumab (Avastin), a monoclonal antibody that binds to VEGF-A and prevents it from activating its receptors. In preclinical mouse models, such VEGF-targeting agents demonstrated remarkable efficacy in slowing or even reversing tumor growth, solidifying the concept of anti-VEGF therapy as a cornerstone of cancer treatment.
However, translating this success from mouse models to human patients proved more complicated. While drugs like bevacizumab have been approved for several cancers, including colorectal, lung, and renal cell carcinoma, the clinical benefits have been modest. For instance, in metastatic colorectal cancer, bevacizumab extended patient survival by only a few months when used with chemotherapy.
Nonetheless, the foundational work on antiangiogenesis remains vital. It not only revolutionized how researchers understand tumor biology but also provided a blueprint for targeting the tumor microenvironment—a strategy still being refined today.
The Clinical Disappointment — Why Anti-VEGF Doesn’t Work as Expected
Despite the initial optimism surrounding anti-VEGF therapies, their performance in clinical oncology has fallen short of expectations. In controlled laboratory settings, antiangiogenic drugs like bevacizumab consistently inhibit tumor growth in mouse models. These successes fueled hopes that blocking VEGF-A signaling would deliver similar benefits in human cancers. However, real-world outcomes have often been modest, raising critical questions about the limitations of this approach.
In human patients, anti-VEGF therapies have demonstrated only incremental improvements in survival, typically just a few months, and often only in combination with chemotherapy. For example, bevacizumab added to irinotecan-based chemotherapy prolonged survival in metastatic colorectal cancer by approximately 4 to 5 months. Moreover, these drugs are not curative and require long-term administration, which can lead to serious side effects, such as hypertension, thromboembolic events, and impaired wound healing.
Why does anti-VEGF therapy fall short in humans? One major reason lies in the maturity and complexity of human tumors. Unlike murine models, where tumors are artificially introduced and treated early, human cancers are often diagnosed at advanced stages, after months or years of progression. By this time, the tumor vasculature has typically evolved to a more resistant state, often supported by pericytes and smooth muscle cells that provide VEGF-independent survival signals.
Another challenge is redundancy in angiogenic signaling. When VEGF is blocked, tumors can compensate by secreting alternative pro-angiogenic factors such as FGF, PDGF, IL-8, and HGF. This biological backup undermines the specificity of anti-VEGF therapies and enables continued vascular growth even in the face of treatment. Additionally, hypoxic conditions induced by therapy can further upregulate VEGF and other survival pathways, reinforcing a feedback loop that supports tumor persistence.
Complicating matters further is the concept of “vascular normalization”, a transient phase during which anti-VEGF therapy reduces vessel leakiness and improves perfusion. While this window can enhance chemotherapy delivery, it also paradoxically improves oxygen and nutrient supply, which may benefit tumor cells .
Thus, while VEGF-targeting drugs remain a key part of the cancer treatment arsenal, their limitations underscore the need for multi-targeted strategies and deeper understanding of tumor vascular biology.
Tumor Vasculature Isn’t One-Size-Fits-All
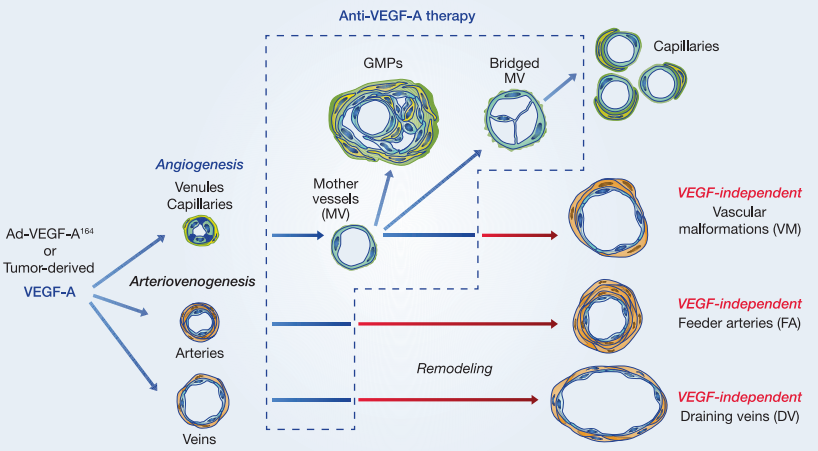
One of the most critical insights emerging from recent research is that tumor blood vessels are highly heterogeneous. Contrary to the early assumption that tumor vasculature forms a uniform network of abnormal capillaries, scientists now understand that tumors are supplied by a diverse array of vessel subtypes, each with distinct structural and functional properties. This diversity plays a significant role in determining a tumor’s response to antiangiogenic therapies such as VEGF inhibitors.
The landmark study by Sitohy et al. (2012) identified six distinct blood vessel types commonly found in both human tumors and mouse models:
Mother Vessels (MVs)
Glomeruloid Microvascular Proliferations (GMPs)
Capillaries
Vascular Malformations (VMs)
Feeder Arteries (FAs)
Draining Veins (DVs)
These vessel types arise through two interconnected biological processes: angiogenesis (formation of new vessels from pre-existing ones) and arterio-venogenesis (remodeling of existing arteries and veins). Initially, anti-VEGF therapy was believed to suppress all tumor vasculature. However, only a subset of these vessels—particularly MVs and GMPs—remain highly dependent on VEGF-A and are therefore sensitive to VEGF-targeting drugs.
As tumors progress, they tend to develop VEGF-independent vessels, such as VMs, FAs, and DVs, which are coated with smooth muscle cells or pericytes. These cells provide alternative survival signals, rendering the vessels resistant to VEGF inhibition. Importantly, these mature vessels become the backbone of the tumor’s blood supply, supporting continued growth even under antiangiogenic treatment.
This heterogeneity poses a major challenge for therapy. By pruning only VEGF-sensitive vessels while sparing the resistant ones, treatments may fail to fully disrupt the tumor’s blood supply. Furthermore, these resistant vessels often appear structurally more “normal,” complicating the interpretation of imaging and treatment outcomes.
Recognizing and targeting this vascular complexity is essential for improving the efficacy of antiangiogenic strategies. It suggests a need to expand therapeutic targets beyond VEGF-A and consider vessel type-specific markers that can more comprehensively shut down tumor perfusion.
What the Mouse Models Reveal About Resistance
Mouse models have been instrumental in helping researchers unravel why many tumors eventually develop resistance to anti-VEGF therapies. One of the most revealing studies used a nonreplicating adenoviral vector expressing VEGF-A164 (the murine equivalent of human VEGF-A165) to mimic tumor-induced angiogenesis in immunodeficient mice. This model allowed scientists to observe, in a controlled environment, how different types of blood vessels develop and respond to VEGF-targeted treatments over time.
When anti-VEGF agents such as rapamycin (an mTOR inhibitor) or aflibercept (VEGF Trap) were administered early—before or soon after the induction of angiogenesis—both drugs effectively regressed immature vessels like mother vessels (MVs) and glomeruloid microvascular proliferations (GMPs). These early vessel types are highly dependent on VEGF-A signaling and are thus vulnerable to its inhibition.
However, a striking pattern emerged when therapy was delayed. If anti-VEGF treatment was initiated after the formation of vascular malformations (VMs), feeder arteries (FAs), and draining veins (DVs), the efficacy of the drugs dropped dramatically. These late-stage, mature vessels remained structurally intact and functionally active, even under aggressive anti-VEGF regimens. Immunohistochemical analysis showed that these vessels expressed minimal to undetectable levels of VEGFR-2, explaining their reduced responsiveness to VEGF blockade.
Further, these mature vessels were coated with smooth muscle cells and pericytes, which are believed to provide alternative growth and survival signals independent of VEGF-A. This structural support makes the vessels more resistant not only to VEGF-targeted drugs but possibly to other stressors such as hypoxia or mechanical damage.
These findings underscore a crucial takeaway: timing matters. Anti-VEGF therapy is most effective during early tumor development when the vasculature is still plastic and VEGF-dependent. Once a tumor has established a mature, VEGF-independent vascular system, these treatments are far less impactful. This helps explain why antiangiogenic drugs show strong effects in young mouse tumors but underperform in late-stage human cancers.
Rethinking the Target — Future of Anti-Vascular Cancer Therapy
As researchers continue to grapple with the limitations of anti-VEGF therapy in treating cancer, a new consensus is emerging: targeting VEGF alone is not enough. While VEGF-A plays a crucial role in initiating angiogenesis, especially in early tumor growth, it is increasingly clear that mature tumor vasculature evolves beyond VEGF dependency. To truly disrupt a tumor’s lifeline, future therapies must expand their scope to include other vascular targets.
One promising avenue is to focus on the larger, VEGF-resistant vessels—specifically vascular malformations, feeder arteries, and draining veins—which remain intact after anti-VEGF treatment. These vessels are often lined by endothelial cells with low VEGFR-2 expression and surrounded by supportive pericytes or smooth muscle cells, making them resistant to current antiangiogenic drugs. Because these structures serve as the main conduits for blood delivery and drainage within tumors, targeting them could disrupt the entire vascular architecture more effectively than pruning VEGF-sensitive microvessels.
To achieve this, researchers are now seeking new molecular markers expressed by these resistant vessels. These markers could serve as entry points for next-generation therapeutics, including antibody-drug conjugates, small-molecule inhibitors, and even photodynamic therapies. For example, experimental approaches using vascular-targeted photodynamic therapy (VTP) have shown success in permanently occluding feeding arteries and draining veins, effectively starving tumors in preclinical models.
Another clinically relevant example is uterine artery embolization for fibroid treatment—a procedure that occludes the blood supply and leads to tissue regression. These non-VEGF-dependent strategies demonstrate that mechanically or functionally disrupting blood flow can yield therapeutic benefits without relying solely on VEGF signaling.
Ultimately, a multi-pronged vascular targeting strategy—one that combines anti-VEGF agents with therapies aimed at mature vasculature—holds the most promise. This approach could overcome resistance, enhance tumor starvation, and improve outcomes across multiple cancer types.
As the field evolves, the focus is shifting from simply blocking angiogenesis to shutting down the tumor’s vascular infrastructure altogether—one major vessel at a time.
References
Folkman, J. (1971). Tumor angiogenesis: therapeutic implications. New England Journal of Medicine, 285(21), 1182–1186.
https://doi.org/10.1056/NEJM197111182852108
Leung, D. W., Cachianes, G., Kuang, W. J., Goeddel, D. V., & Ferrara, N. (1989). Vascular endothelial growth factor is a secreted angiogenic mitogen. Science, 246(4935), 1306–1309.
https://doi.org/10.1126/science.2479986
Ferrara, N., Hillan, K. J., & Novotny, W. (2004). Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochemical and Biophysical Research Communications, 333(2), 328–335.
https://doi.org/10.1016/j.bbrc.2005.05.132
Hurwitz, H., Fehrenbacher, L., Novotny, W., Cartwright, T., Hainsworth, J., Heim, W., … & Kabbinavar, F. (2004). Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. New England Journal of Medicine, 350(23), 2335–2342.
https://doi.org/10.1056/NEJMoa032691
Bergers, G., & Hanahan, D. (2008). Modes of resistance to anti-angiogenic therapy. Nature Reviews Cancer, 8(8), 592–603.
https://doi.org/10.1038/nrc2442
Carmeliet, P., & Jain, R. K. (2011). Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nature Reviews Drug Discovery, 10(6), 417–427.
https://doi.org/10.1038/nrd3455
Nagy, J. A., Chang, S. H., Shih, S. C., Dvorak, A. M., & Dvorak, H. F. (2010). Heterogeneity of the tumor vasculature. Seminars in Thrombosis and Hemostasis, 36(3), 321–331.
https://doi.org/10.1055/s-0030-1253454
Nagy, J. A., Dvorak, A. M., & Dvorak, H. F. (2007). VEGF-A and the induction of pathological angiogenesis. Annual Review of Pathology: Mechanisms of Disease, 2, 251–275.
https://doi.org/10.1146/annurev.pathol.2.010506.134925
Sitohy, B., Nagy, J. A., & Dvorak, H. F. (2012). Anti-VEGF/VEGFR therapy for cancer: Reassessing the target. Cancer Research, 72(8), 1909–1914.
https://doi.org/10.1158/0008-5472.CAN-11-3406
Sitohy, B., Nagy, J. A., Jaminet, S. C., & Dvorak, H. F. (2011). Tumor-surrogate blood vessel subtypes exhibit differential susceptibility to anti-VEGF therapy. Cancer Research, 71(20), 7021–7028.
https://doi.org/10.1158/0008-5472.CAN-11-0761
Xue, Q., Nagy, J. A., Manseau, E. J., Phung, T. L., Dvorak, H. F., & Benjamin, L. E. (2009). Rapamycin inhibition of the Akt/mTOR pathway blocks select stages of VEGF-A164-driven angiogenesis, in part by blocking S6Kinase. Arteriosclerosis, Thrombosis, and Vascular Biology, 29(8), 1172–1178.
https://doi.org/10.1161/ATVBAHA.109.188276
Evensen, L., Micklem, D. R., Blois, A., Berge, S. V., Aarsaether, N., Littlewood-Evans, A., et al. (2009). Mural cell associated VEGF is required for organotypic vessel formation. PLoS ONE, 4(6), e5798.
https://doi.org/10.1371/journal.pone.0005798
Madar-Balakirski, N., Tempel-Brami, C., Kalchenko, V., Brenner, O., Varon, D., Scherz, A., et al. (2010). Permanent occlusion of feeding arteries and draining veins in solid mouse tumors by vascular targeted photodynamic therapy (VTP) with Tookad. PLoS ONE, 5(8), e10282.
https://doi.org/10.1371/journal.pone.0010282
Levy, B. S. (2008). Modern management of uterine fibroids. Acta Obstetricia et Gynecologica Scandinavica, 87(8), 812–823.
https://doi.org/10.1080/00016340802215226
Seaman, S., Stevens, J., Yang, M. Y., Logsdon, D., Graff-Cherry, C., & St. Croix, B. (2007). Genes that distinguish physiological and pathological angiogenesis. Cancer Cell, 11(6), 539–554.