AKT and Cancer: Unlocking the Master Switch Behind Tumor Growth and Therapy Resistance
Abstract
The AKT signaling pathway is a pivotal regulator of cellular survival, proliferation, and metabolism, making it one of the most studied drivers of tumorigenesis. This blog post explores the origins, structure, and activation mechanisms of AKT isoforms (AKT1, AKT2, AKT3), their diverse oncogenic functions, and their contributions to key cancer hallmarks such as apoptosis resistance, cell cycle progression, and angiogenesis. Drawing on the influential commentary by Testa and Bellacosa (2001), we examine how AKT affects subcellular protein localization and how this contributes to malignancy. The article also discusses current clinical implications, therapeutic challenges, and future directions in targeting the AKT pathway for cancer treatment. Understanding the full scope of AKT’s role in cancer is essential for developing precise and durable anti-cancer therapies.
Why AKT Matters in Cancer Biology
In the complex world of cancer biology, signaling pathways are the invisible conductors that determine whether a cell grows, survives, or dies. Among them, the AKT pathway has emerged as one of the most influential players in tumorigenesis. AKT, also known as protein kinase B (PKB), is not a single protein but a family of kinases—AKT1, AKT2, and AKT3—that serve as central nodes in a vast web of intracellular signals. These kinases integrate cues from growth factors, hormones, and nutrients, translating them into powerful cellular responses.
What makes AKT especially critical is its capacity to regulate multiple hallmarks of cancer, including resistance to apoptosis, sustained proliferation, and enhanced metabolism. In other words, when the AKT pathway is deregulated—as often occurs in various malignancies—it can tip the balance toward unchecked tumor growth. Genetic alterations such as overexpression, amplification, or hyperactivation of AKT isoforms have been observed in several cancers, including ovarian, breast, prostate, and pancreatic cancers.
The paper by Testa and Bellacosa (2001) was instrumental in highlighting the central role of AKT in cancer. It synthesized years of research to illustrate how AKT not only supports cell survival but also interferes with tumor suppressor functions, modulates transcription factors, and facilitates cell cycle progression. Their insights helped reframe AKT from being a simple downstream kinase to a “master regulator” of oncogenic processes.
Today, AKT is recognized not just as a biomarker for aggressive cancers but also as a promising therapeutic target. The pathway’s importance spans both basic research and clinical oncology, making it essential knowledge for anyone interested in understanding or combating cancer. As our molecular toolkit evolves, so too does the potential for targeting AKT in increasingly precise and effective ways.
The AKT Family – Isoforms, Origins, and Activation Mechanism
The AKT family of kinases—AKT1, AKT2, and AKT3—serves as a central conduit for intracellular signaling with profound effects on cell survival, metabolism, and proliferation. First discovered through studies on transforming retroviruses, the v-akt oncogene was isolated from a mouse T-cell lymphoma, encoding a serine/threonine kinase fused to viral sequences that conferred constitutive activation. This landmark finding paved the way for identifying human homologues, each encoded by separate genes on chromosomes 14q32 (AKT1), 19q13 (AKT2), and 1q44 (AKT3).
Despite their structural similarity, the AKT isoforms exhibit distinct expression profiles and physiological roles. AKT2, for example, is highly expressed in insulin-responsive tissues like adipose and liver. Its knockout in mice leads to insulin resistance and impaired glucose homeostasis, underscoring its role in metabolic regulation. Meanwhile, AKT3 appears to be more abundant in the brain and may contribute to neurodevelopment and hormone-insensitive cancers. These isoform-specific nuances suggest functional divergence, although considerable redundancy still exists in their oncogenic capacity.
AKT activation is tightly regulated through a multi-step process initiated by receptor tyrosine kinases (RTKs) and mediated by phosphoinositide 3-kinase (PI3K). Upon activation, PI3K catalyzes the production of phosphatidylinositol-3,4,5-trisphosphate (PIP3), which recruits AKT to the plasma membrane via its pleckstrin homology (PH) domain. Once localized, AKT is phosphorylated at two key residues: Thr308 by PDK1 and Ser473 by the elusive PDK2 complex, resulting in full enzymatic activation.
This tightly orchestrated cascade can be disrupted in cancer. One of the most common mutations involves the PTEN tumor suppressor, which dephosphorylates PIP3, thus inhibiting AKT activation. Loss of PTEN function leads to uncontrolled AKT signaling, promoting oncogenic processes across multiple tumor types. This mechanistic clarity has driven the development of AKT and PI3K inhibitors as targeted therapies.
Oncogenic Functions – How AKT Fuels Tumor Growth and Survival
Once activated, AKT exerts powerful control over cell fate, acting as a central node in promoting survival, proliferation, and metabolic adaptation. Its oncogenic potency lies in its ability to simultaneously manipulate several molecular processes that drive tumor progression. One of AKT’s most studied roles is its anti-apoptotic function, where it blocks programmed cell death by targeting key molecules. For instance, AKT phosphorylates and inactivates the pro-apoptotic proteins BAD and procaspase-9, preventing mitochondrial cytochrome c release and caspase cascade activation. It also activates NF-κB signaling through IκB kinase (IKK), driving the transcription of anti-apoptotic genes.
Beyond survival, AKT facilitates cell cycle progression by downregulating inhibitors and stabilizing growth-promoting factors. It promotes the cytoplasmic retention of p21^WAF1^ and p27^Kip1^, two cyclin-dependent kinase inhibitors, thereby impeding their ability to halt the cell cycle in the nucleus. AKT also enhances the stability of cyclins D1 and D3 and supports mRNA translation by phosphorylating 4E-BP1, allowing eIF4E to initiate protein synthesis—processes crucial for rapid tumor cell growth.
AKT’s influence extends to angiogenesis and replicative immortality, two additional hallmarks of cancer. It activates endothelial nitric oxide synthase (eNOS), stimulating vascular remodeling and new blood vessel formation, which tumors need to grow. Simultaneously, AKT enhances telomerase activity via phosphorylation of human telomerase reverse transcriptase (hTERT), contributing to the unlimited division potential of cancer cells.
Recent studies also highlight AKT’s role in nuclear-cytoplasmic shuttling of key regulators. For example, it enables Mdm2 nuclear entry, which downregulates p53, a critical tumor suppressor. In doing so, AKT helps cancer cells bypass genomic safeguards and continue proliferating under conditions that would normally trigger arrest or apoptosis.
Together, these interconnected functions make AKT a formidable oncogenic driver—capable of tipping cellular equilibrium in favor of malignancy.
Clinical Implications – From Cancer Hallmarks to Targeted Therapy
The widespread influence of AKT signaling across multiple cancer hallmarks has made it a prime target in the search for new cancer therapies. According to Hanahan and Weinberg’s seminal framework, the hallmarks of cancer include traits such as resistance to cell death, sustained proliferative signaling, angiogenesis, and invasion—all processes in which AKT plays a direct or supporting role. By promoting survival signals, destabilizing tumor suppressors, and enhancing cell motility, AKT signaling equips cancer cells with the molecular tools to grow uncontrollably and spread.
In particular, AKT2 amplification and overexpression are frequently observed in ovarian, pancreatic, and breast cancers, while AKT1 activation is associated with poor prognosis in prostate and gastric tumors. AKT3, although less commonly amplified, shows elevated activity in estrogen receptor-negative breast cancers and androgen-independent prostate cancer, suggesting a role in aggressive, hormone-refractory disease subtypes.
Clinically, the implications are profound. Tumors exhibiting PI3K/AKT pathway dysregulation often respond poorly to standard therapies, as AKT can mediate both chemo- and radio-resistance by blocking apoptosis and promoting DNA repair mechanisms. This resistance has prompted intensive efforts to develop AKT inhibitors, some of which have entered clinical trials. These include both pan-AKT inhibitors and isoform-specific compounds designed to reduce off-target effects.
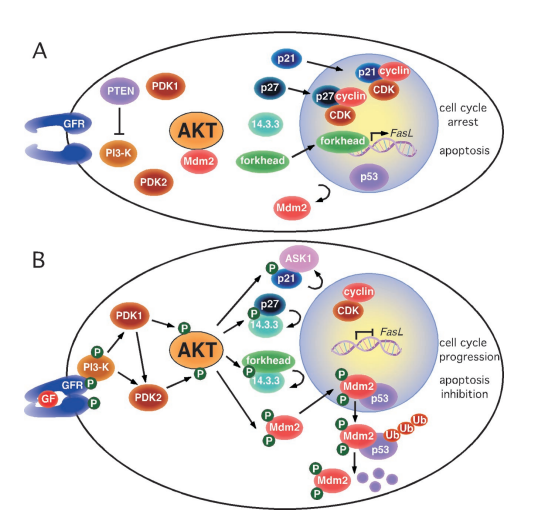
Fig. 1. Phosphorylation by AKT regulates compartmentalization of multiple substrates involved in cell cycle progression and inhibition of apoptosis.
However, targeting AKT is not without challenges. Feedback loops, pathway redundancy, and compensatory signaling can diminish drug efficacy. For instance, inhibition of AKT may activate upstream receptor tyrosine kinases or shift signaling through alternative pathways like MAPK. As such, combination therapies—pairing AKT inhibitors with other agents targeting mTOR, HER2, or MEK—are being actively explored to overcome resistance.
Ultimately, the AKT pathway is more than a therapeutic target—it is a molecular signature of tumor adaptability and aggressiveness. A deeper understanding of its role in cancer subtypes and treatment resistance will be essential for designing next-generation targeted therapies.
Future Directions – Why the AKT Pathway Still Deserves Attention
More than two decades after its pivotal recognition in cancer biology, the AKT pathway remains at the forefront of oncological research and drug development. The enduring interest in AKT stems not only from its involvement in nearly every hallmark of cancer but also from its resilience as a therapeutic challenge. While multiple inhibitors targeting PI3K, AKT, and downstream effectors like mTOR have been developed, achieving durable clinical responses has proven complex due to signaling redundancy and feedback activation.
Future research is expected to focus on several promising directions. One area is the isoform-specific targeting of AKT1, AKT2, and AKT3. Although these isoforms share structural similarities, emerging evidence suggests they may govern distinct biological outcomes in different tissues or cancer subtypes. For instance, AKT1 tends to be more involved in proliferation, while AKT2 plays a critical role in metastasis and metabolic regulation. Understanding these nuances could lead to more selective inhibitors with fewer side effects.
Another key frontier is the interactome mapping of AKT substrates. As noted in Testa and Bellacosa’s commentary, AKT affects cellular fate not just through enzyme activity but also by controlling the subcellular localization of proteins such as Mdm2, p21, p27, and forkhead transcription factors. Dissecting how AKT modulates these interactions at the structural and regulatory level may reveal new therapeutic vulnerabilities.
There is also increasing interest in combinatorial treatment strategies. Rather than targeting AKT in isolation, researchers are exploring combinations with immunotherapies, DNA damage response inhibitors, or epigenetic drugs to counteract resistance mechanisms. The rationale is to exploit AKT’s integration point within broader signaling networks to maximize therapeutic outcomes.
Finally, as technologies like CRISPR, phosphoproteomics, and single-cell sequencing evolve, they promise to uncover patient-specific insights into AKT signaling alterations—advancing the dream of truly personalized cancer therapy.
References
Bellacosa, A., Testa, J. R., Staal, S. P., & Tsichlis, P. N. (1991). A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science, 254(5029), 274–277.
https://doi.org/10.1126/science.1925581
Cheng, J. Q., Godwin, A. K., Bellacosa, A., Taguchi, T., Franke, T. F., Hamilton, T. C., … & Testa, J. R. (1992). AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proceedings of the National Academy of Sciences, 89(19), 9267–9271.
https://doi.org/10.1073/pnas.89.19.9267
Testa, J. R., & Bellacosa, A. (2001). AKT plays a central role in tumorigenesis. Proceedings of the National Academy of Sciences, 98(20), 10983–10985.
https://doi.org/10.1073/pnas.211430998
Hanahan, D., & Weinberg, R. A. (2000). The hallmarks of cancer. Cell, 100(1), 57–70.
https://doi.org/10.1016/S0092-8674(00)81683-9
Cho, H., Mu, J., Kim, J. K., Thorvaldsen, J. L., Chu, Q., Crenshaw, E. B., … & Birnbaum, M. J. (2001). Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science, 292(5522), 1728–1731.
https://doi.org/10.1126/science.292.5522.1728
Di Cristofano, A., & Pandolfi, P. P. (2000). The multiple roles of PTEN in tumor suppression. Cell, 100(4), 387–390.
https://doi.org/10.1016/S0092-8674(00)80674-1
Zhou, B. P., Liao, Y., Xia, W., Spohn, B., Lee, M. H., & Hung, M. C. (2001). Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nature Cell Biology, 3(3), 245–252.
https://doi.org/10.1038/35060098
Kang, S. S., Kwon, T., Kwon, D. Y., & Do, S. I. (1999). Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. Journal of Biological Chemistry, 274(19), 13085–13090.
https://doi.org/10.1074/jbc.274.19.13085
Datta, S. R., Brunet, A., & Greenberg, M. E. (1999). Cellular survival: A play in three Akts. Genes & Development, 13(22), 2905–2927.
https://doi.org/10.1101/gad.13.22.2905
Cheng, J. Q., Ruggeri, B., Klein, W. M., Sonoda, G., Altomare, D. A., Watson, D. K., & Testa, J. R. (1996). Amplification and expression of AKT2 in human pancreatic cancer cells and their inhibition by antisense RNA. Proceedings of the National Academy of Sciences, 93(8), 3636–3641.
https://doi.org/10.1073/pnas.93.8.3636
Nakatani, K., Thompson, D. A., Barthel, A., Sakaue, H., Liu, W., Weigel, R. J., & Roth, R. A. (1999). Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. Journal of Biological Chemistry, 274(31), 21528–21532.
https://doi.org/10.1074/jbc.274.31.21528
Sun, M., Wang, G., Paciga, J. E., Feldman, R. I., Yuan, Z. Q., Ma, X. L., … & Cheng, J. Q. (2001). AKT1 and AKT2 protein kinases are frequently hyperactivated in human prostate and breast cancers. American Journal of Pathology, 159(2), 431–437.
https://doi.org/10.1016/S0002-9440(10)61720-2
Mende, I., Malstrom, S., Tsichlis, P. N., Vogt, P. K., & Aoki, M. (2001). Oncogenic transformation induced by membrane-targeted Akt2 and Akt3. Oncogene, 20(35), 4419–4423.
https://doi.org/10.1038/sj.onc.1204573
Alessi, D. R., & Cohen, P. (1998). Mechanism of activation and function of protein kinase B. Current Opinion in Genetics & Development, 8(1), 55–62.