MUSE
CHEM
RC Products
Featured Chemicals
Bioactive Chemicals
Buliding Block Chemicals
Catalysts and Ligands
Materials Science
Chemical Reagents
Reference Standards
Inhibitors/Agonists
Service
Antibody-drug conjugates
Bioconjugation-Service
Analytical Services
Custom Synthesis
Peptide Service
Order Center
Distributors
Terms
About
About us
Citations
Blog
CONTACT
You are searching: I013052
Chemical Reagents
Catalysts and Ligands
Bioactive Chemicals
Apoptosis
Load More
View All
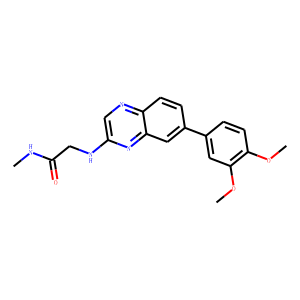
BQR-695
Cat No. I013052
CAS Number: 1513879-21-4
Purity: ≥ 95%
Discover
1